Abstract
The PrecisionFDA recently organized the Truth Challenge V2 to compare the accuracy of mapping and variant calling algorithms in difficult-to-map regions relying on new truth sets derived from linked and long-read sequencing. The PrecisionFDA announced the challenge results on August 3rd and the DRAGEN mapper + variant caller won the accuracy contest for Illumina reads in the Difficult-to-Map Regions and All Benchmark Regions categories, with 38% and 28% fewer call errors than the second-best contestants, respectively. DRAGEN performance in this challenge beat the previous release of DRAGEN by almost 50%, while still analyzing the whole genome in less than 25 minutes on a single 20 core server +1 FPGA. To achieve these results, DRAGEN alt-aware mapping capability was used, adding several hundred thousand short, alternate contigs derived from phased population haplotypes to the hg38 reference, effectively evolving it towards a graph reference. Additionally, the DRAGEN variant caller made use of new techniques to better identify and handle overlapping variants in a read pile-up.
Introduction
An estimated 99.9% of the human genome is conserved across individuals. That means that it is only 0.1% that contributes to a person’s elevated cancer risk, predicts their inability to metabolize a specific drug, or explains the etiology of their heritable disease. Accurately identifying the genetic variants between individuals is essential in precision medicine, disease diagnosis, research into genetic origin of disorders and drug discovery. To that extent the Genome In a Bottle (GiaB) consortium and National Institute of Standards and Technology (NIST) have provided truth sets of select subjects such as NA12878, covering a significant portion of the genome, allowing bioinformaticians to measure the accuracy of their tools in identifying variants. Of particular importance are the mapping and variant calling steps.
The GiAB consortium and NIST recently used linked and long reads to release an expanded truth set, V4.2, that covers 92% of the genome (up from the previous 85%), with significantly improved coverage of homologous regions and difficult-to-map-regions, which include 193 clinically relevant genes. These truth sets are an invaluable resource and key enabler to develop and benchmark secondary analysis methods into previously more opaque regions of the genome. While the NIST truth sets were developed by leveraging long-read technology to characterize regions that are typically difficult to access, we demonstrate here that via breakthrough improvements to both DRAGEN mapping capabilities and small variant calling, DRAGEN now offers much better accuracy in difficult-to-map regions of the genome with Illumina reads. These results highlight two key takeaways:
- By leveraging population haplotypes of phased variants and augmenting the reference index with population derived alt contigs, the DRAGEN mapper can effectively map against a graph genome and improve the mapping of Illumina reads in those difficult regions. This new feature effectively extends the reach of Illumina reads and enables accurate mapping and variant calling in regions that could not be accessed before.
- What makes this achievement particularly important is that the difficult-to-map regions contain 193 clinically relevant genes for which variants could not be easily identified without a specific targeted assay and specialized informatics. With these recent improvements, DRAGEN can now yield much better variant calling accuracy in these genes from a normal WGS/WES run, which in turn leads to better diagnostics in clinical labs.
Illumina reads are by far the most widely used sequencing technology because of their accuracy and cost-effectiveness. Enabling short reads to resolve difficult-to-map regions of the genome, which contain many clinically relevant genes, provides the opportunity to further accelerate diagnostic decisions and continue to help identify disease-causing variants in patients.
PrecisionFDA challenge overview
The PrecisionFDA Truth Challenge V2 was sponsored by PrecisionFDA and the GIAB consortium with NIST. This challenge was launched to assess small variant calling pipeline performance on a common frame of reference, with a focus on benchmarking in difficult-to-map regions, segmental duplications, and the Major Histocompatibility Complex (MHC).
The Genome in a Bottle (GIAB) consortium recently used linked and long reads to develop an expanded set of high-confidence truth calls for a specific trio. High quality long reads typically yield higher confidence calls in difficult-to-map regions. With that capability, the GIAB expanded truth set now covers 7% of the genome beyond prior truth sets, including many medically relevant genes. The extended truth regions have over 270 million bases in low-mappability regions and segmental duplications.1
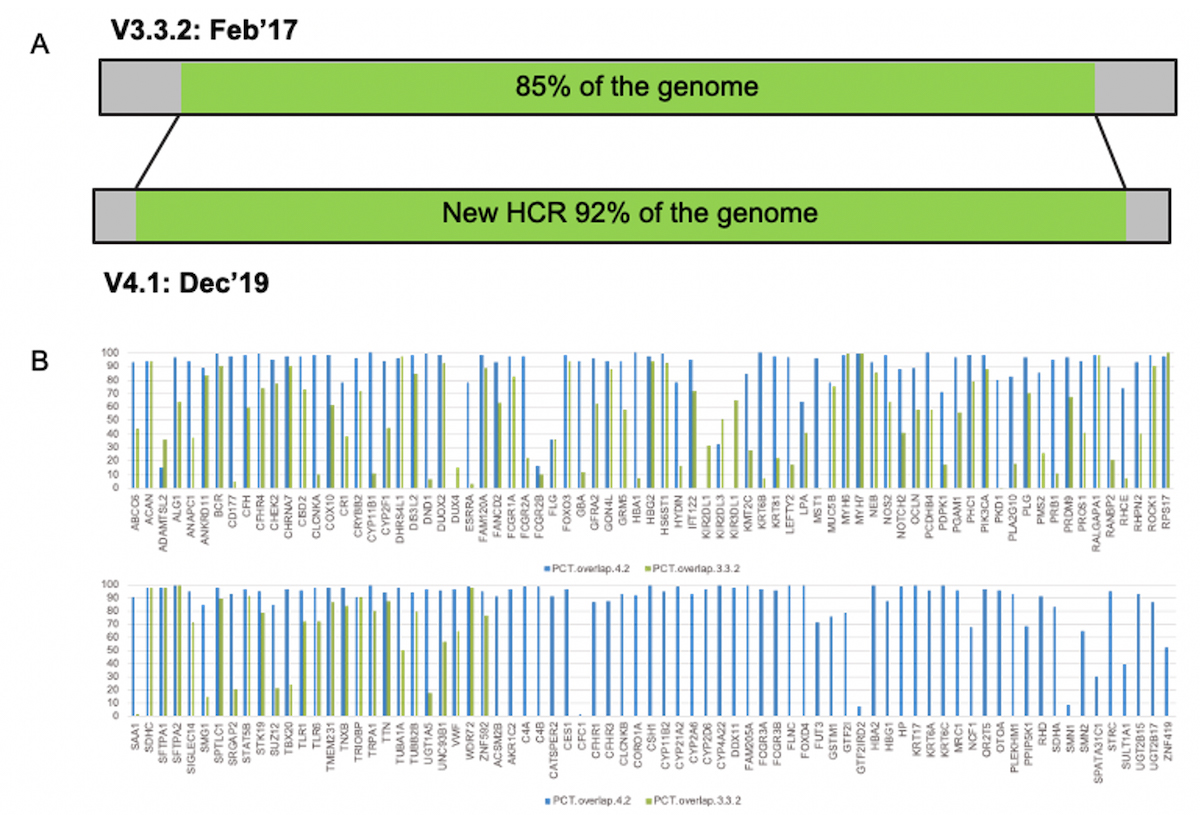
A) genome covered by the high confidence truth set increased from 85% to 92%. B) percentage coverage comparison between previous and expanded truth sets, for 193 clinically-relevant genes. Blue bars show the coverage % with the extended truth set.
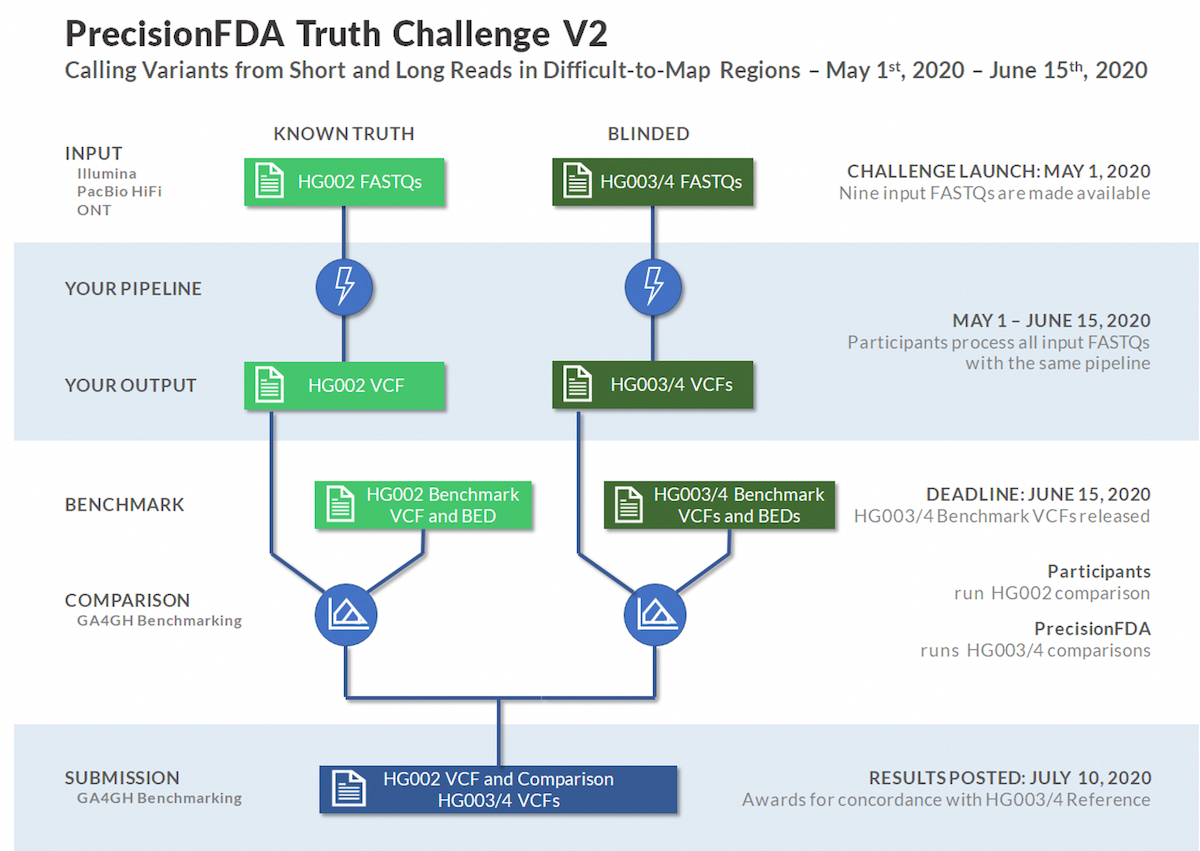
Source: https://precision.fda.gov/challenges/10
PrecisionFDA challenge results
DRAGEN competed in the Illumina reads category and ranked in first in two of the three test regions (Difficult-to-Map Regions, and All Benchmark Regions). In Figure 3, the bars show the total number of errors for SNV and INDEL combined, FP+FN, on the y-axis and the various submissions results on the x-axis. The DRAGEN submission won by a comfortable margin with around 28% and 38% fewer errors compared to the second-best submission, in the All Benchmark Regions and Difficult-to-Map Regions respectively.
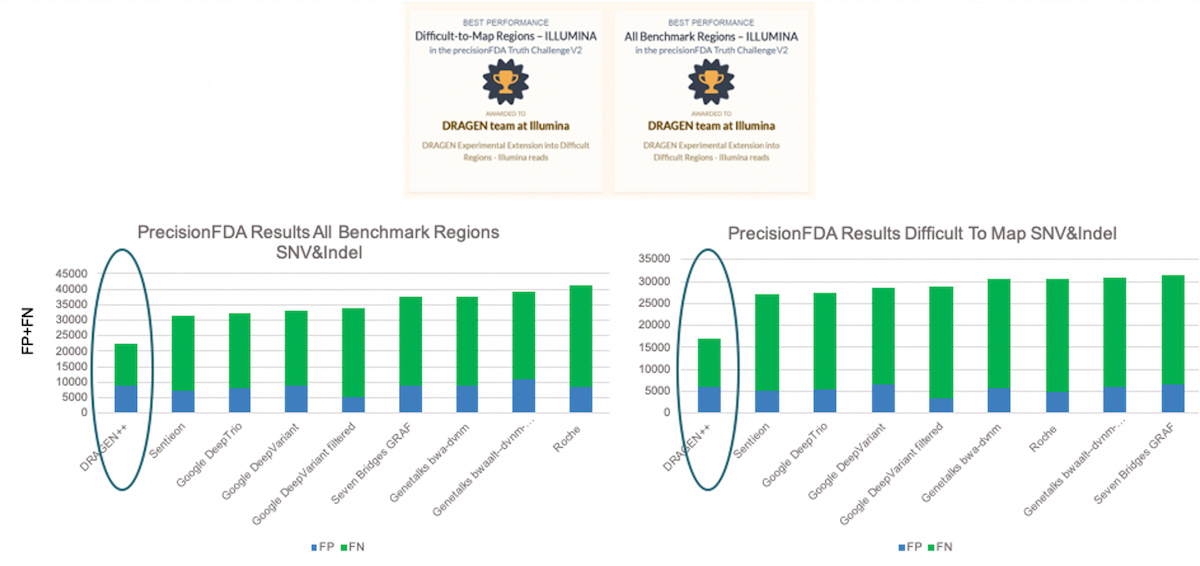
DRAGEN submissions won best performance awards in two of the three test regions (Difficult-to-Map Regions, and All Benchmark Regions) on Illumina sequencing data with a comfortable margin compared to the other submissions.
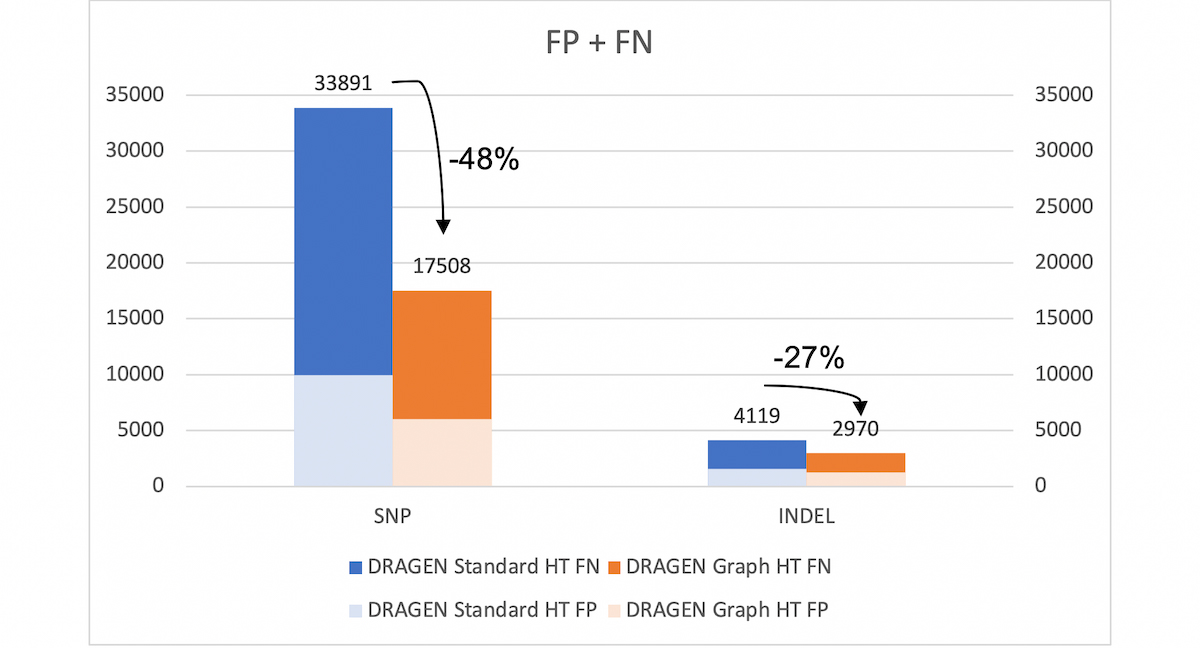
With the extended v.4.2 truth set (VCF and BED), DRAGEN graph reduces the SNP errors by ~48% and INDEL errors by ~27% compared to legacy DRAGEN. *HT: hash-table
Important note to users
The 50% reduction in total errors from DRAGEN graph is measurable with the extended truth set v4.2. This performance gain is not apparent when benchmarking against the older v3.3.2 truth set. That is partly because the v3.3.2 truth set does not include the difficult to map regions, but also because the v4.2 truth set corrects errors that were present in v3.3.2. Indeed, DRAGEN graph appears to yield additional SNP and INDEL false positives (FP) compared to legacy DRAGEN when using the older truth set v3.3.2. However, a large proportion of the additional FP calls are due to the v3.3.2 truth VCF being incomplete. The same ‘FP’ variants are marked as true positives in the v4.2 truth VCF.
To show that DRAGEN graph effectively extends the reach of Illumina reads, we compare the DRAGEN accuracy in the extended truth V4.2 truth set against that achievable by PacBio HiFi long reads with DeepVariant caller.
The PacBio HiFi reads made available as part of the PrecisionFDA Truth Challenge V22 were aligned using PBMM2 v1.3 and processed using DeepVariant v1.0.0 and WhatsHap v1.0, exactly as outlined in the deepvariant-pacbio-model-case-study found in.3 This case study involves a two-step process of variant calling. After the first round of calling, SNVs are phased and used to haplotag the input BAM. The haplotagged BAM is then used as input to DeepVariant again, variants are called, and are phased once more. The final phase 2 VCF was used for all comparisons.
As shown in Figure 5, DRAGEN graph gets significantly closer to the SNP accuracy of long reads than the previous DRAGEN version. Both versions of DRAGEN yield better INDEL accuracy than the long reads pipeline.
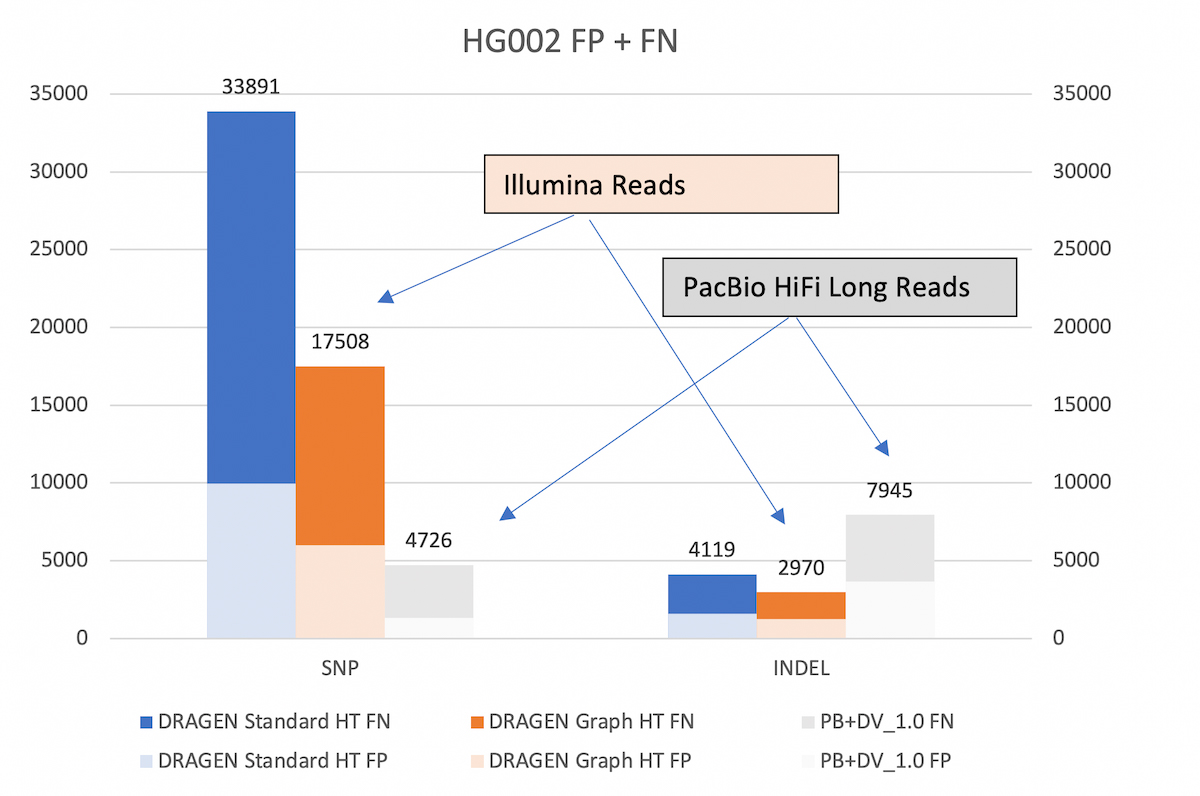
Legacy DRAGEN vs DRAGEN Graph vs. PacBio+DV
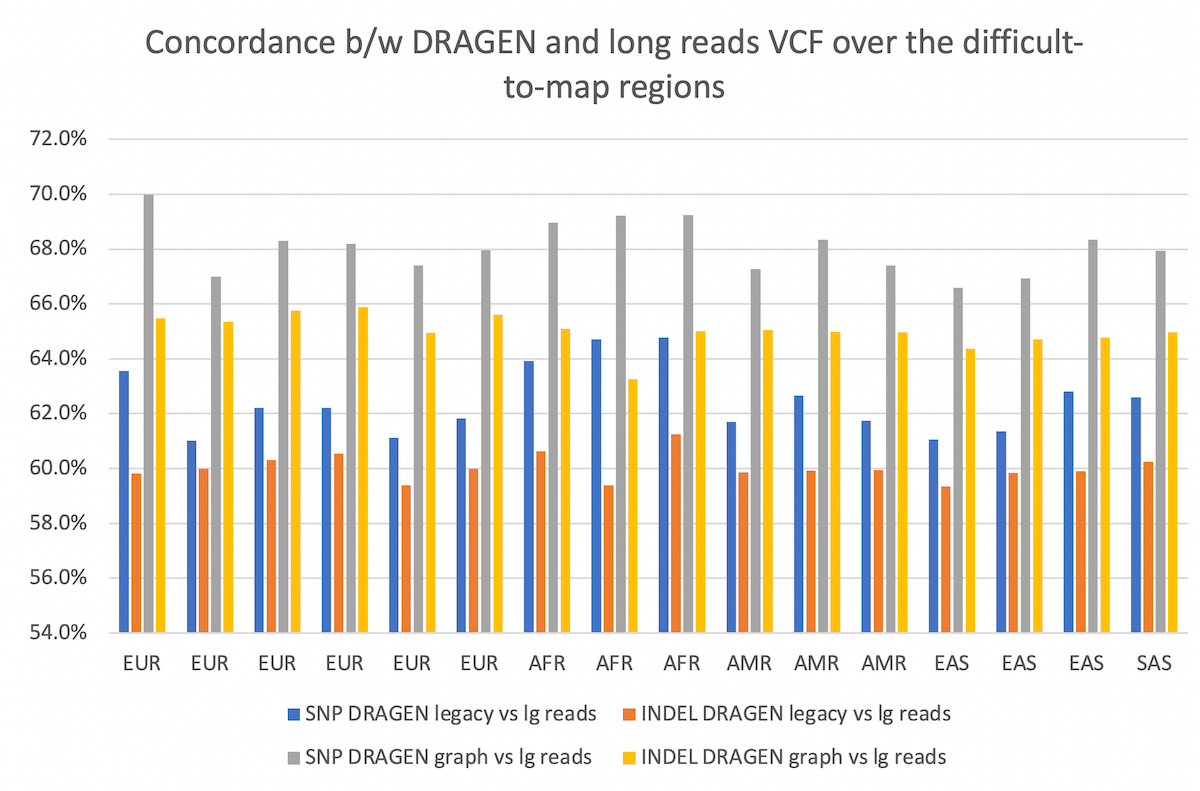
The comparison is collected over the difficult-to-map regions. The concordance between the two pipelines is improved by using DRAGEN graph, by ~5-6% for both SNP and INDEL, and preserved across all populations considered.
DRAGEN graph: backwards compatibility and run times
Besides the massive reduction in total number of errors in the extended truth set, the other attractive point of the DRAGEN graph is that it is fully compatible with standard BAM and VCF file formats, and with existing reference genomes. To enable DRAGEN graph, the user just needs to update the hash table with the graph hash-table (HT) and this can be done via DRAGEN command line. Mapping with this HT yields a standard hg38 BAM, with graph alignments automatically projected to the primary assembly. Currently the DRAGEN graph HT is available in the hg38 domain but a version for the GRCh37 domain will soon be available. What’s more is that the graph capability comes at no extra cost in run time.
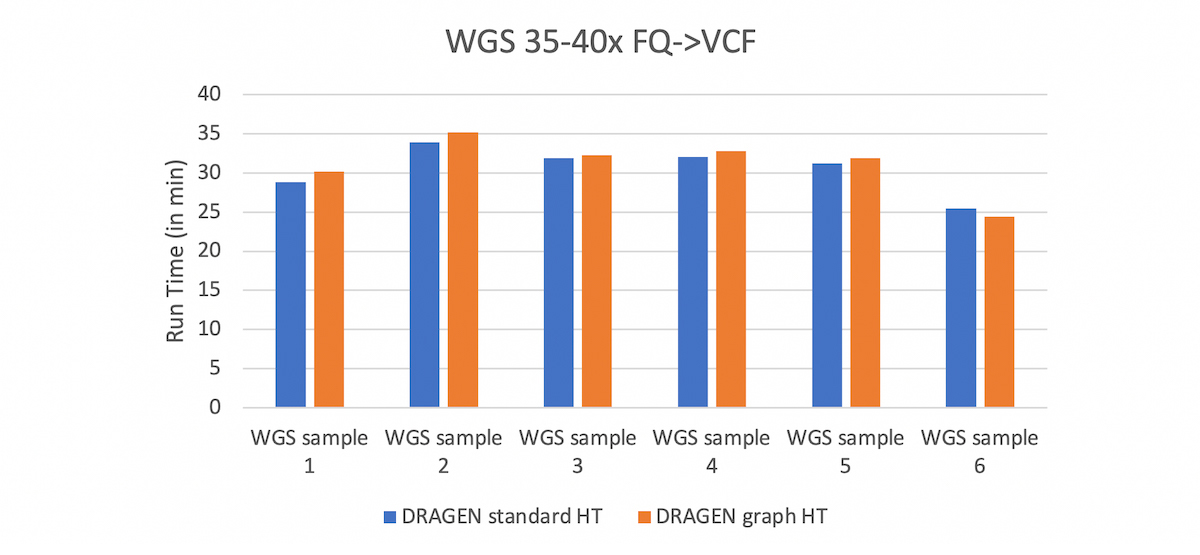
Comparison between DRAGEN graph HT and DRAGEN Standard HT. The run times are very comparable which shows that adding the graph genome capability comes at no cost in run time.
Next, we do a deeper dive into the new DRAGEN mapping capabilities and Variant Calling improvements that produced the winning accuracy results in the PrecisionFDA Truth V2 challenge.
DRAGEN innovations: support for graph genomes
The PrecisionFDA Truth V2 challenge focused on "difficult-to-map" regions, the primary regions where the GIAB Consortium expanded their benchmarks. Accurate variant calling with short-read data is quite challenging and error-prone in these regions. Perhaps unsurprisingly, the main impediment to accurate analysis in "difficult-to-map" regions is difficulty in mapping short reads into these regions accurately. A variant caller analyzes the pileup of reads mapped to a given locus to determine the most probable original sequence content there, but can’t do this accurately if the pileup is missing many of the evidence reads that should be present, or is contaminated with mis-mapped foreign reads, or alignments with minimal mapping confidence (MAPQ).
Mapping difficulty can arise because a region (such as MHC) is highly polymorphic, and sample reads differ so greatly from the reference genome that a mapper can’t find or recognize a good match. Much more commonly, though, mapping difficulty arises when sample reads match a region reasonably well – but also match other regions almost or just as well. This happens when near copies of a region appear in several places in the reference genome (segmental duplicates), or in the case of common highly repetitive sequences.
In many cases, such mapping difficulty can be overcome by utilizing known variation patterns in the population, rather than just a single reference genome. Suppose a short read (or read pair) matches two regions, A and B, equally well but imperfectly, with two nucleotide differences from the reference genome in each region. Based on the reference alone, a mapper can only pick A or B at random and align there with zero MAPQ. But suppose we know that the read’s two differences from region A occur commonly in the population, while its two differences from B have not been observed in the population. We can use this knowledge as a guide to map the read to region A, with reasonably high confidence.
A graph reference is an approach to aiding mapping with population data which has long been proposed. In a graph reference, alternate sequence content observed in the population is represented as various diverging and converging paths. Figure 8A shows how several types of variation can be represented. Sample reads may be allowed to align to any best-matching path through the reference graph.
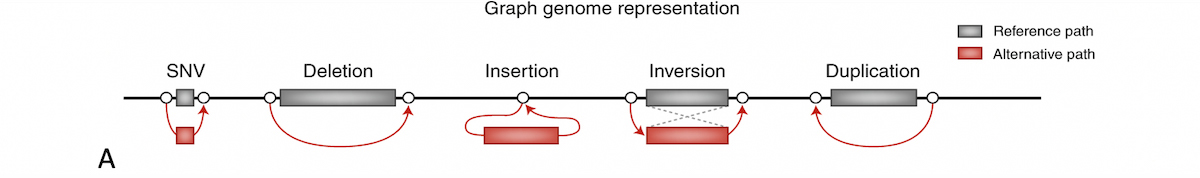
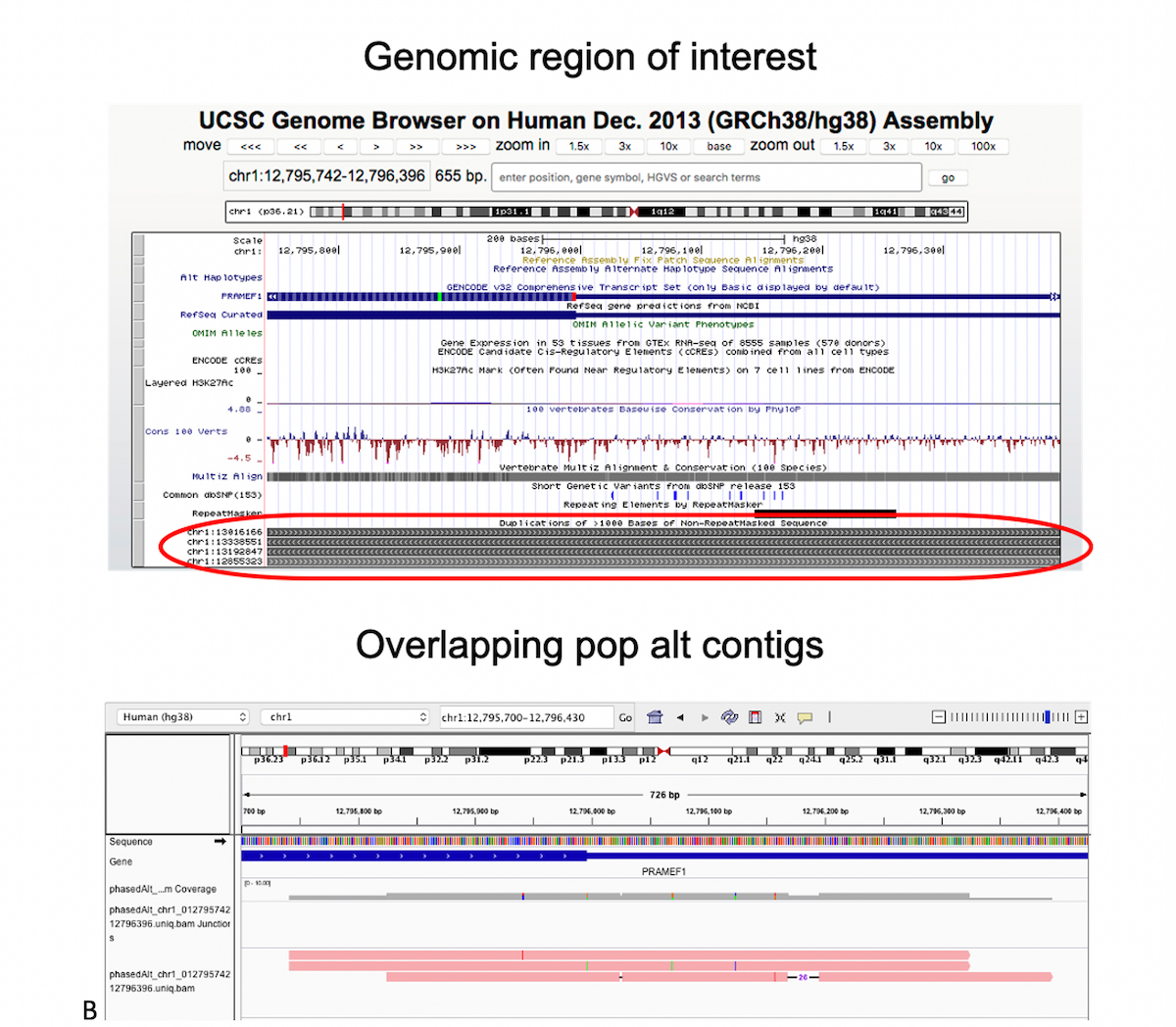
A) Graph genome overview: In a graph reference, alternate sequence content observed in the population is represented as various diverging and converging paths. B) Example of multiple phased alt contigs overlapping a given region with segmental duplication.
The DRAGEN Mapper, like other commonly used mappers, uses a linear reference as a baseline. But it has two capabilities which support augmenting the linear reference into an effective graph, and mapping reads in a manner identical to first aligning them to the implicit graph, then projecting those alignments to the linear reference path.
First, the DRAGEN Mapper supports injecting a population nucleotide substitution as a multi-base IUPAC-IUB code in the reference sequence. For example, a reference “A” nucleotide known to sometimes be “G” in the population can be encoded as “R”. When aligning a read across the “R” position, an “A” or “G” in the read both score as a match. This is identical behavior to aligning a read to the equivalent graph construct, a linear path which forks into “A” and “G” paths then immediately converges again. We also upgraded DRAGEN seed mapping to populate seed K-mers matching either the reference or alternate base into the mapping hash table, so there is no sensitivity loss mapping to such positions.
Second, the DRAGEN Mapper has an advanced “alt awareness” capability which allows augmenting the reference with “alt contigs” representing alternate paths in a graph, each with predefined liftover alignments into the linear reference. Whenever sample reads align best to one of the alt contigs, the liftover knowledge is used to report an alignment to the projected position in the linear reference – with potentially high confidence (MAPQ) because the alt contig alignment is not treated as competing with its linear reference projection, but rather as a guide to that projected alignment. Once again, this is identical behavior to aligning reads to an equivalent graph construct. But in this case, any graph-reference construct can theoretically be represented as one or more alt contigs with appropriate liftover.
The DRAGEN team used these two capabilities to augment hg38 with observed population small variants in the difficult-to-map regions. For this challenge, we limited the population sources to phased variants called from 16 long-read (PacBio HiFi) European samples. We injected isolated population nucleotide substitutions (SNVs) as multi-base codes, and more complex population variation as added alt contigs: insertions and deletions, complex substitutions, and clusters of small variants phased on a single haplotype.
Figure 8B shows a Chromosome 1 region which is classified difficult-to-map due to several other segmental duplicate copies, as seen at the bottom of the UCSC browser view. We added three alt contigs over this region, the pink tracks at the bottom of Figure 7B, each containing a small number of phased small variants. Just these few population landmarks can be sufficient to guide many reads to the correct segmental duplicate copy.
Having thus augmented the reference with the population variants, the beauty of our approach was that mapping operated normally, as if the reference was just hg38. The DRAGEN Mapper did the work of using the multi-base codes and alt contigs to improve alignment scores where sample reads matched population haplotypes, and of projecting the best alignments onto the linear hg38 reference for use in variant calling. Mapping to the augmented reference simply yielded a more accurate hg38 BAM, which dramatically improved variant calling accuracy without modification to the variant caller.
To verify that the DRAGEN graph accuracy gains observed for the Ashkenazi trio generalize to other individuals, we needed to find a method that does not rely on truth sets, as NIST v4.2 truth sets are currently available only for the Ashkenazi trio HG002/3/4. Instead, we randomly selected 9 participants from the European-descent cohort in the 1000 Genomes Project, and compared aligned reads to the reference with DRAGEN legacy and DRAGEN graph. We found that DRAGEN graph substantially reduces the base substitution rate (mismatch rate between aligned read bases and corresponding reference sequence) across all samples analyzed (Figure 9. The base substitution rate of aligned reads vs. GRCh38 is shown on the left, and DRAGEN_GRCh38_graph on the right. None of the 9 individuals were used in the construction of the graph reference. The base substitution rates were calculated for the portion of GRCh38 spanned by the phased haplotypes (about 110Mb). A reduced base substitution rate indicates improved read mapping, which can enable better variant calling accuracy. This result demonstrates that the gain of DRAGEN graph extends to other individuals.
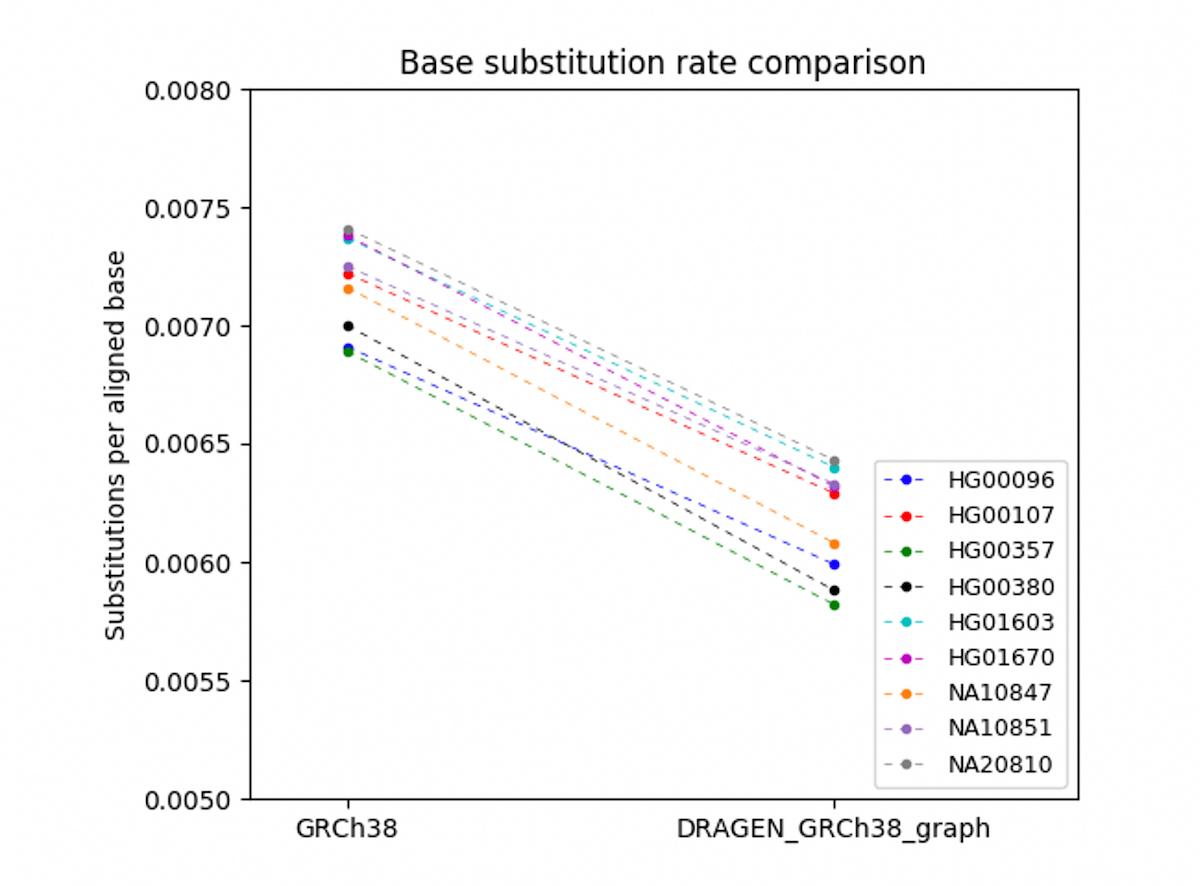
Comparison between legacy GRCh38 Hash Table and 'DRAGEN_GRCh38_graph' which uses the hg38 hash table augmented with the pop alt contigs.
We also observed that DRAGEN graph may correct reference bias. Figure 10 shows the Variant Allele Frequency (VAF) distribution of the heterozygous variant calls from the VCF obtained with DRAGEN legacy and with DRAGEN graph, intersected with the low-mappability bed. Over the low mappability regions, the graph-based distributions are closer to what is expected in theory (ie, the graph VAF distribution is better centered and more symmetric around 0.5).
One could conjecture that the graph skews the VAF distribution higher, because the alt contigs act more strongly to pull reads containing alt alleles in a given position into their correct pileup, than to pull in ref-supporting reads. But since the distribution is now more centered around 50%, the biased action of the alt contigs is really a corrective. It also stands to reason that, in the absence of alt contigs, at a difficult-to-map locus, reads supporting an alt allele are more likely to be missing from the pileup, or have very low MAPQ. So we hypothesize that the main impact of the alt contigs in this regard is to mitigate reference bias and therefore correct the VAF distribution.
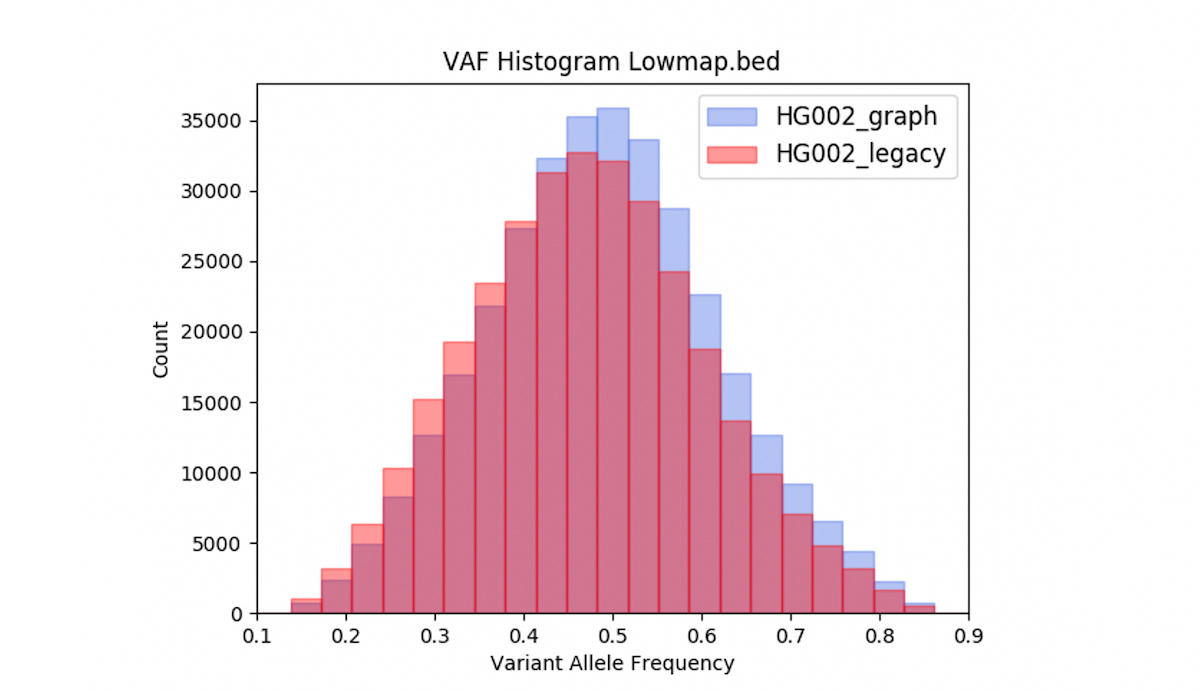
Graph mode corrects reference bias, and shifts the VAF mean, median, and skewness statistics closer to their predicted values.
DRAGEN innovations: joint detection of overlapping variants
For additional accuracy gains, we tackled one more low-hanging fruit in small variant calling. The genotyping calculation in conventional variant callers is designed to consider a single locus at a time when genotyping events. This performs well where events are separated and independent but fails to accurately call overlapping events (between SNP and INDELs), or events separated by a short tandem repeat (STR) region. We hypothesized that we could improve variant calling accuracy by merging nearby loci into a single region and genotyping pairs of haplotypes, instead of pairs of events.
We identify regions for joint detection (JD) of multiple variants at multiple loci using the following criteria: loci have alleles which overlap each other, loci are in a STR region or less than 10 bases away from an STR region, or loci are less than 10 bases away from each other. STR regions are good candidates for joint detection because 1) this is where PCR-induced INDEL errors occur, which may overlap with true variant SNP, 2) this is also where true INDEL variants occur, which may overlap among each other or with SNP, 3) there are situations where a homozygous INDEL has half of its reads misaligned to represent the INDEL at the end of a homopolymer rather than at its true location (eg, beginning or middle of a homopolymer). JD is effective at recovering the true variants in all these cases.
We then modify the variant caller algorithm to generate a complete haplotype list where all possible combinations of the alleles within the JD region are represented. Then, during the genotyping step, we calculate the likelihood of each haplotype pair given the observed read pileup. Finally, using a haplotype-to-allele mapping, we calculate genotype posterior probabilities for pairs of events and report the genotype with the maximum posterior probability.
We found that this led to substantial reductions in false negatives, most notably in INDELs (Figure 11). JD helps rescue variant calls that previously went undetected (because of too low confidence score), and it also helps correct genotype errors (eg, converting heterozygous calls into homozygous calls), both of which improves sensitivity. The reason why JD especially benefits INDELs is because it resolves situations involving INDEL (INDEL overlapping SNP, INDEL in STR regions), which were previously not handled properly via a simple column-wise based genotyping of events.
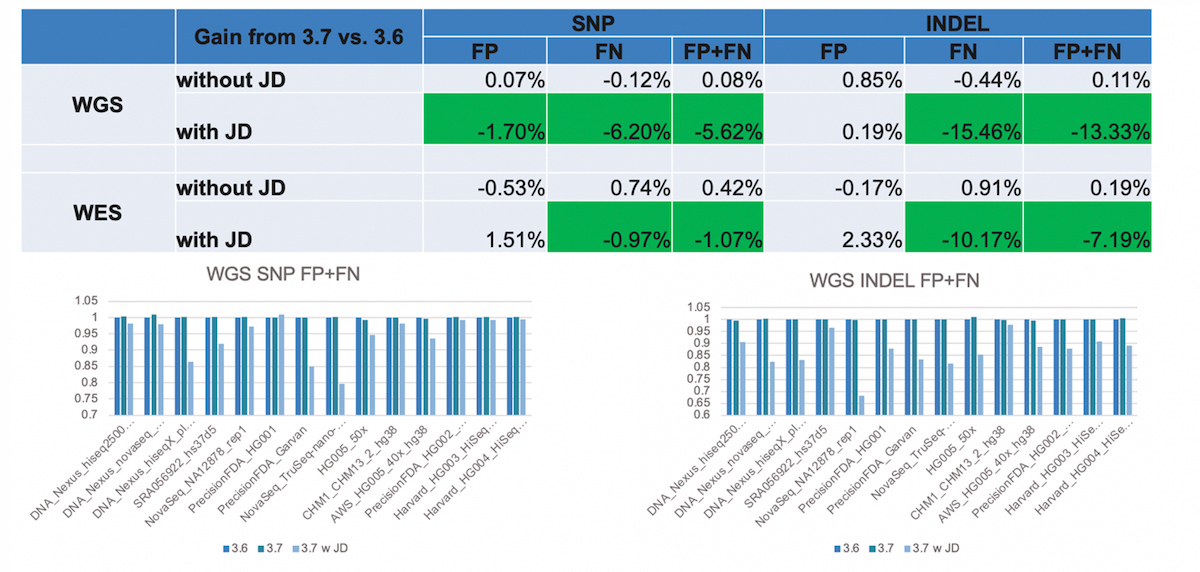
- Significant improvement in FP+FN with JD, esp. for INDELs. Gain in sensitivity.
- Bar charts show a comparison of DRAGEN v.3.6, v.3.7 with and without JD.
DRAGEN graph significantly reduces total errors count in the extended truth set
Figure 12 shows that with conventional pipelines (DRAGEN 3.6 and BWA-MEM), the total number of errors increases significantly when switching over from the previous truth set (v.3.3.2) to the extended truth set (v.4.1). The increase in total errors is due to added FN of variants present in the extended truth sets in regions that are not easily accessible to conventional short-reads pipelines. With DRAGEN graph (shown as DRAGEN 3.7), the total number of errors FP+FN is significantly reduced.
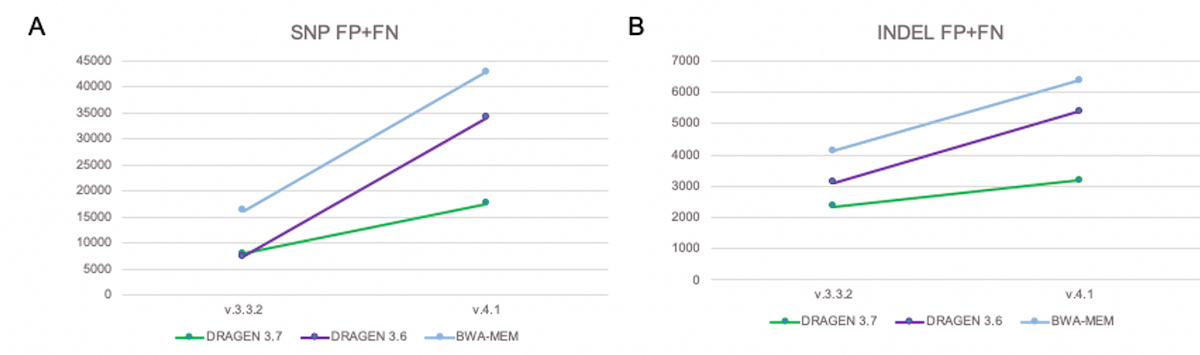
A) SNP FP+FN B) INDEL FP+FN
Summary of results & applicability of results beyond PrecisionFDA challenge
The challenge offered an opportunity for the participants in general and certainly for Illumina's DRAGEN team to focus on new areas of innovation, the fruit of which will come out over the next few releases of DRAGEN starting with DRAGEN 3.7 released on October 26, 2020. We see the methods used for this challenge as a start of major new capabilities in DRAGEN that will be improved and generalized to include structural variants, copy number variations and repeat expansions. DRAGEN Graph can represent haplotypes of multiple ancestries in its reference, improving accuracy across the board and mitigating biases from a linear reference. These capabilities have already shown that even with 2 × 150 long read pairs, we can now call variants with much better accuracy than what was thought possible in difficult to map regions. Equipped with this experience, we will relentlessly strive to perfect and extend the analysis in the remaining regions.