DNAシーケンスと下流データ処理のコストが低下するにつれて、集団シーケンス研究は前例のない規模で実行可能になりつつあります。コホートレベルのバリエーションカタログは、祖先研究、希少バリアント解析、および遺伝型/表現型の関連性を探索するための重要なリソースです。そのため、コールセットが高精度であることは重要ですが、データ処理と解析の課題が残ったままです。
大規模コホートからのコールセットを集積するためのツールがいくつかあります。その一例として、GATKとその共通ジェノタイピングワークフローがあり1、長年にわたってスタンダードとなっていました。しかし、GATKによってコールされるバリアントセットに限り必要とされる手順がいくつか含まれています。これらは、バリアントスコア再キャリブレーション(VQSR)などのその他のバリアントコーラーには適用されません。他にも、DNAnexusが開発し、クラウドプラットフォームに密接に結合されているGLnexus2があります。このコーラーで他のクラウドプラットフォームやHPCに大規模に展開する方法は明らかではありません。多くの既存ツールに存在するもう1つの課題は、効率的な反復解析であり、実現すれば、既存のコールセットに新しいサンプルとそのバリアントを追加することができます。
DRAGEN gVCF Genotyperは、集団規模で小さな生殖細胞系列バリアントを集積し、ジェノタイピングするイルミナのソリューションです。gVCF Genotyperへのインプットは、DRAGEN生殖細胞系列バリアントコーラーによって書き込まれるgVCFファイル3のセットです。アウトプットは、コホートで発見されたすべてのバリアントの遺伝型を含むマルチサンプルVCFファイル(msVCF)です。アウトプットには、品質管理やバリアントフィルタリングに使用できるコホートメトリクスも含まれます。
DRAGEN gVCF Genotyperは、既存のコホートに新しいサンプルを追加するための反復ワークフローを実装しています。このワークフローにより、ユーザーは処理を繰り返すことなく、サンプルの新しいバッチを既存のバッチと効率的に組み合わせることができます。
ユーザーはアウトプットmsVCFをHail(https://hail.is)やPLINK(https://www.cog-genomics.org/plink)などの下流ツールに取り込んで、データ探索や関連解析を行うことができます。
この記事では、gVCF Genotyperワークフローの概要の説明、このワークフローを大規模に導入する方法が紹介されています。また、1000 Genomes Projectコホート(1KGP)を使用したDRAGEN gVCF Genotyperの性能を実証しています。
当社ではこれまでに、いくつかの社内プロジェクトでgVCF Genotyperを使用してきました。例えば、DRAGEN 4.0および最大50万のサンプルを持つ大規模コホートを使用した1KGPコホートの再解析(https://www.internationalgenome.org)などです。外部ユーザーには、Genomics England社4,5などが含まれています。
このソフトウェアは、次世代シーケンサーデータ解析のためのイルミナのソリューション、DRAGENの一部です。DRAGEN 4.0では、gVCF Genotyperは分散処理モードを使用する場合、数十万のサンプルに拡張できます。gVCF Genotyperを実行する際に推奨される最もスケーラブルなプラットフォームはIllumina Connected Analytics(ICA)ですが、イルミナではDRAGENサーバーを使用したオンプレミス解析やHPCでの展開もサポートしています。
大規模なジェノタイピング
DRAGEN gVCF Genotyperは、VCFと同様にバリアント情報と特定の位置(ホモ接合性リファレンスの位置)に存在しないバリアントの信頼度の両方を含むgVCFインプット形式に依存します。
ジェノタイピングワークフローはいくつかのステップで構成されています。まず、各入力ファイルからバリアントアリルを抽出します。バリアントジェノタイプやその他のメトリクスを保存します。次のステップでは、特定位置の全アリルをキャプチャーするバリアント部位を収集します。低品質のアレルと、コールされた遺伝型でサポートされないアリルは、フィルターを適用して廃棄します。これには、この位置のホモ接合性リファレンスであるサンプルからの情報を使用することが含まれます。
その後、バリアントアリルがノーマライズされます。インプットにあるアリルのインデックスからアリルマップを計算してその部位のアリル配列へとマッピングします。以下に例を示します(表1)。ノーマライズされたアリルのセットと一致するように、各サンプルの遺伝型とQCメトリクスも更新します。

スケーラブルなVCF表現
gVCF Genotyperのアウトプットは、コホート内のすべてのサンプルに対して計算されたメトリクスを含むmsVCFです。
コホートサイズが大きくなると、msVCFは大きくなる可能性があります。場合によっては、VCFパーサーが割り当てるよりも多くのメモリが必要になるため、メモリとランタイムに課題が生じます。これは、FORMAT/PLなどのアリルの組み合わせの可能性ごとに値を保存するVCFエントリによって引き起こされます。したがって、アリルの数に応じて四分の一にスケーリングします。ゲノムの複雑性が低い領域では、1部位あたりのアリル数が多い可能性があります。また、サンプル数とともに大幅に増加します。
これらのすべての理由から、FORMAT/PLをFORMAT/LPLタグに置き換えました。これにより、サンプルで発生するアリルの値のみを保存し、コホート内のこの部位で見つかったすべてのアレルの保存はしません。また、各サンプルに新しいFORMAT/LAAフィールドを追加しました。このフィールドには、アリルのグローバルリストとは異なり、このサンプルで発生する代替対立遺伝子の1ベースのインデックスがリストアップされます。
このアプローチはローカルアリルと呼ばれ、BCFtools(https://samtools.github.io/bcftools/bcftools.html)やHail(https://hail.is/docs/0.2/experimental/vcf_combiner.html#local-alleles)などのオープンソースソフトウェアでも使用されています。
msVCFには、各アリルのアリル数と近交係数も含まれます。アリルカウントはアリル頻度の計算に使用されます。これは特に、バリアントが良性である可能性が高いか、または病原性である可能性があるかを判断するための有用なメトリクスです。
近交係数(IC)は、バリアント部位における過剰なヘテロ接合性を測定します。また、リードマッピングアーティファクトの検出とフィルタリングにも使用できます。過剰ヘテロ接合性が高いゲノムの6遺伝子座は、実際にはゲノムの異なる部位に属するリードがあるため、ヘテロ接合性のように見える不正確なリードマッピングの領域である可能性があります。
近交係数は、1 −(観察されたヘテロ接合性遺伝型の数)/(予想されるヘテロ接合性遺伝型の数)として算出します。この数値が負の場合、Hardy-Weinbergの仮定(https://en.wikipedia.org/wiki/Hardy%E2%80%93Weinberg_principle)で想定したよりも多くのヘテロ接合体が観察されていることを意味します。

良い経験則は、負の育種係数でバリアント部位をフィルタリングすることです。6 図1は、これが有用なアプローチである理由を示しています。負の育種係数は、コールレート(NS_GT)などの他のフィルターでフィルタリングされない高いヘテロ接合性を持つ部位をキャプチャします。
以下は、DRAGEN gVCF Genotyperによって書き込まれたmsVCFファイルの図で、前述のとおり「local alleles」形式を使用しています(図2)。

GATKのようなオープンソースソフトウェアは、genotype likelihood(FORMAT/PL)のフルセットを書き込んでいますが、DRAGEN gVCF GenotyperやHailのようなその他のツールは、サンプルで実際に発生するアリル(「ローカルアリル」)のみのセットを書き込んでいます。
GATKのベストプラクティスとの比較
GATK(Genome Analysis Toolkit)のベストプラクティスでは、クオリティースコアの再キャリブレーション、データベースへの取り込み、遺伝型の精緻化(ジョイントジェノタイピングとも呼ばれる)など、大規模コホートからのバリアントコールを分析するためのステップをいくつか推奨しています。
ジョイントジェノタイピングは、ジェノタイピングの精度を向上させるためにコホート情報を活用するアルゴリズムのクラスを指します。GATKチームはこの手法のパイオニアでした。
ジェノタイピング精度の向上は重要ですが、前述のとおり7、DRAGENバリアントコールでは精度の向上につながらないため、ジョイントジェノタイピング用のGATKスタイルのアルゴリズムは不要になります。その理由は、GATKアルゴリズムがバリアントのアーティファクトの除去を試みるためですが、DRAGENではこれらは上流ですでにフィルタリングされています。
GATKジョイントジェノタイピングアルゴリズムも計算上高価な操作であるため、DRAGENバリアントコールではGATKスタイルのジョイントジェノタイピングを行わずにDRAGEN gVCF Genotyperのみを実行することを推奨します。
同じことがVQSRにも当てはまります。VQSRは、最新バージョンのDRAGEN(https://community.illumina.com/s/question/0D53l00006lvBDBCA2/why-vqsr-was-removed-from-latest-versions-of-dragen)を使用するバリアントコールセットには推奨されなくなりました。
シングルサーバー解析と分散処理の比較
gVCF Genotyperワークフローは、複数のサンプルバッチが利用可能な場合はステップバイステップモードで、単一のサンプルバッチのみが利用可能な場合はシングルサーバー解析モードで実行できます。
gVCF Genotyperに使用されるFASTAリファレンスは、アライメントおよびバリアントコール用のDRAGENハッシュテーブルの構築に使用されるものと同じでなければなりません。
シングルサーバー解析モードは、一連のgVCFインプットファイルを1つのmsVCFに集約します。限られたサンプル数について、メトリクスと遺伝型を用いてmsVCFを迅速に生成すると便利です。このモードでは、反復解析はできません。
シングルサーバー解析ワークフローの詳細は、DRAGEN 4.0ユーザーガイドに記載されています8。

図3に示されているステップバイステップモードは、複数のノードにコンピューティングを分散させるいくつかのステップから構成されています。ワークフローは数十万のサンプルまで拡張できます。
ステップ1(gVCFの集約): gVCFファイルのバッチをコホートファイルと全数調査ファイルに集約します。コホートファイルは、マルチサンプルgVCFと同様に、複数のサンプルからのgVCFデータを保存するための要約データ形式となっています。全数調査ファイルは、コホート内の各サンプルについて、ホモ接合性リファレンス遺伝型の全バリアントおよびブロックの要約統計量を保存します。多くのサンプルが処理されている場合、ユーザーはそれらを同じサイズの複数のバッチ(例:1000サンプル)に分割し、各バッチでステップ1を別々に実行する必要があります。
ステップ2(全数調査の集計): すべてのバッチの全数調査ファイルが生成された後、ユーザーはそれらを単一のグローバル全数調査ファイルに統合できます。このステップは数千バッチに拡大されます。
ステップ3(msVCF生成):グローバルの国勢調査ファイルが生成されると、ユーザーはバッチごとのコホートファイルと国勢調査ファイルとともにこれを使用して、サンプルのバッチごとにmsVCFファイルを生成できます。
複数のコンピューティングノードで並列処理を容易にするために、上記のすべてのステップで、ユーザーはゲノムを同じサイズの断片に分割し(コマンドラインオプションを使用 - ユーザーガイドで説明されている断片)、異なるコンピューティングノードで反復gVCF Genotyperの1つのインスタンスを使用して各断片を処理することができます。ゲノムの断片は染色体の境界で破壊され、断片の数に下限を課します。
このワークフローでは、(サンプルバッチ数)*(ゲノム断片数)msVCF出力ファイルという結果が得られます。すべてのバッチのmsVCFを1つにまとめる必要がある場合は、すべてのバッチのmsVCFファイルを、すべてのサンプルが含まれる1つのmsVCFへと統合するために、追加ステップを実行できます。
サンプルの新しいバッチが利用可能になったら、ユーザーはそのバッチにステップ1を実行すればよく、その後、新しいバッチの全数調査ファイルを以前のすべてのバッチのグローバル全数調査ファイルと組み合わせて、新しいグローバル全数調査ファイルを書き込みます。新しいバリアント部位が発見されて、グローバル全数調査ファイルが更新されるたびに、および/または既存のバリアント部位でバリアント統計が更新されるたびに、新しい情報を含む新しいバッチ単位のmsVCFファイルを生成できます。
下の図4は、既存のコホートに新しいサンプルバッチを追加するために必要なステップを示しています。新しいバッチのgVCFファイルについては、ステップ1のみを実行して、新しいバッチごとのコホートと全数調査ファイルを作成する必要があります。その後、ワークフローはステップ2を実行し、ステップ3の各バッチのmsVCFファイルを更新するために必要な新しいグローバル調査ファイルを作成します。

クラウドとオンプレミスでの利用可能範囲
DRAGEN gVCF GenotyperはDRAGEN 4.0の一部であり、その他のすべてのコンポーネントと同様にDRAGENサーバーで実行できます。ただし、これは小規模なコホートに対してのみ実行可能です。シングルサーバー解析モードでは、1000gVCFファイルの処理に約24時間かかります。
gVCF Genotyperを反復的に実行するワークフローがICAに実装されており、大規模コホート(10,000サンプル超)のユーザーに対して推奨されるオプションです。ICAワークフローは、100,000を超える生殖系列サンプルでテストされています。
gVCF GenotyperはICAプラットフォームから入手できます。ICA v2では、コマンドラインインターフェース(CLI)を利用できます11。
ICAでgVCF Genotyperを実行するコストは、gVCFからmsVCFまでのシングルサーバー解析ワークフローで1サンプルあたり約0.3 iCreditsです。反復解析のコストは、新規サンプルの追加ごとに約0.3 iCredits、すでに処理済みの各サンプルのmsVCFの更新は0.06 iCreditsです。この推定値は、500コアで実行されるDRAGEN 4.0とAWS地域us-east-1で実行されるICAに基づいています。
ほとんどの場合、このプラットフォームでは、ユーザーはコストとランタイムをトレードオフできます。たとえば、ICAノードの数を減らすとコストは低くなりますが、結果が出るまでの時間は長くなります。
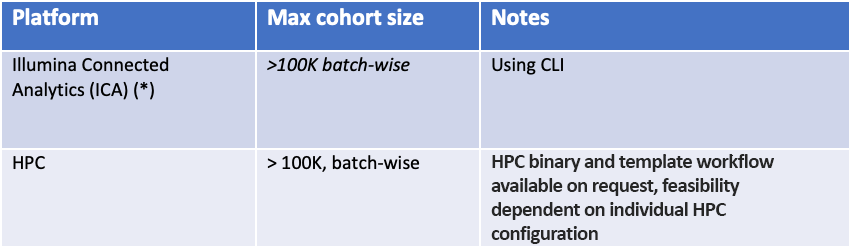
また、コモディティハードウェアやHPC上で実行できるgVCF Genotyperのソフトウェアモードバイナリも提供しています。
gVCF GenotyperのランタイムはディスクI/Oの速度に依存します。入力ファイルのサイズも重要な要素です。
gVCFジェノタイピングプロジェクトを計画する場合は、DRAGEN生殖細胞系列バリアントコーラー(コマンドラインオプション:--vc-compact-gvcf)でコンパクトなgVCFアウトプットモードを使用することを検討してください。これにより、gVCFファイルは40%~50%小さくなり、gVCF Genotyperなどのファイルを解析するソフトウェアで大幅な高速化を実現できます。また、保管コストも削減されます。このコンパクトなgVCFモードは、血統におけるバリアントのde novo検出にのみ必要なgVCFからのメトリクスをいくつか省略します。つまり、コンパクトなgVCFはDRAGEN血統コーラーには使用できないことを意味します。
ユースケース:ICAにおけるThe 1000 Genomes Project解析
7年前、1KGPはローカバレッジシーケンスで構成されるオープンアクセスデータセットを公開しました(26集団の2,504人の無関係な個人のデータ)。これは、ヒトの遺伝的バリエーションのカタログをまとめるための最初の大規模シーケンスの取り組みでした。
ニューヨークゲノムセンターの研究者は、他の科学パートナーらと協力して、多くの親子トリオを追加するため、このリソースを拡大しました。拡大されたセットは、602トリオを含む3,202サンプルで構成され、公開されています9。
ニューヨークゲノムセンターはGATKを使用してこのコホートのバリアントをコールしました。イルミナはDRAGEN 4.0を使用してこれらのサンプルを再解析し、公開しました。このデータリリースについては、別のブログ記事で説明します。
DRAGEN gVCF Genotyperを使用して、3,202サンプルすべてを集約し、ジェノタイピングを実施しました。解析費用は990 iCreditで、サンプルあたり0.31 iCreditに相当します。この解析の結果は、コホート内のすべてのバリアントの遺伝型と頻度としてmsVCFファイル1つに収まりました。また、遺伝型の欠失やHardy-Weinbergの仮定からの逸脱に関する統計も収集し、近交係数で示しました(図1)。
ジェノタイピングされたmsVCFの汎用性と精度を2つの例を用いて示します。
隠れたトリオにおけるメンデルの法則に一貫性のない遺伝型
コホート10の一部であるサンプルNA20891、NA20882、NA20900の隠れたトリオでメンデルエラー率を計算しました。血統関係から逸脱するジェノタイプコールのカウントは、ゲノムの高信頼度領域に限定されないため、精度を評価する上で有用なメトリクスです。
以下の表3は、3人組のうちの1人以上がバリアントを持つ部位の合計数におけるメンデルエラー数を示しています。この表では、GATKコールセットとDRAGEN 4.0および3.5.7で生成されたコールセットを比較しています。

1KGPにおける推定細胞株アーティファクトの解析
ニューヨークゲノムセンターは、コホート全体で1回のみ発生するバリアント(シングルトン、アリルカウント [AC]=1)を解析しました9。これらのバリアントの多くは細胞株のアーティファクトであるという仮説を立てました。また、サンプル中のシングルトン数とコホート内の親族数を、親族がいないサンプル中のシングルトン数と比較しました。小児のシングルトン数は最も少なく、親がその次に少なく、コホートに親戚がいないサンプルがこれに続きました。

DRAGEN gVCF Genotyperは、アリル数などの多くのバリアントメトリクスをその場で計算します。したがって、シングルトンバリアントはその他のツールに頼ることなく容易に抽出および解析できます。上記の図5は、サンプルごとのシングルトンバリアントの内訳を示しています。このプロットは、同系性のステータスに応じてサンプルがどのように分離されるかを示しています。子供はシングルトンが最も少なく、親と他人のサンプルがそれに続く傾向があります。
学術用途向けの詳細情報またはDRAGEN試用版ライセンスについては、dragen-info@illumina.comまでお問い合わせください。
注釈
1. Derek Caetano-Anolles (2022) “Calling variants on cohorts of samples using the HaplotypeCaller in GVCF mode” https://gatk.broadinstitute.org/hc/en-us/articles/360035890411-Calling-variants-on-cohorts-of-samples-using-the-HaplotypeCaller-in-GVCF-mode
2. Michsel F Lin (2018) “GLnexus: joint variant calling for large cohort sequencing “ https://www.biorxiv.org/content/10.1101/343970v1
3. Derek Caetano-Anolles (2022) gVCF – Genomic Variant Call Format https://gatk.broadinstitute.org/hc/en-us/articles/360035531812-GVCF-Genomic-Variant-Call-Format
4. Kousathanas et al (2022) “Whole-genome sequencing reveals host factors underlying critical COVID-19” https://www.nature.com/articles/s41586-022-04576-6 Nature volume 607, pages 97–103
5. Genomics England press release on covid 19 patient study https://www.genomicsengland.co.uk/news/study-insights-severe-covid-19
6. Derek Caetano-Anolles (2022) “Inbreeding coefficient” https://gatk.broadinstitute.org/hc/en-us/articles/360035531992-Inbreeding-Coefficient
7. Population genetics data processing with the DRAGENTM Bio-IT Platform https://jp.support.illumina.com/content/dam/illumina/gcs/assembled-assets/marketing-literature/dragen-popgen-tech-note-m-gl-00561/dragen-popgen-tech-note-m-gl-00561.pdf
8. Instructions for using the Illumina DRAGEN Bio-IT Platform https://support-docs.illumina.com/SW/DRAGEN_v40/Content/SW/DRAGEN/gVCFGenotyper.htm
9. Marta Byrska-Bishop et al (2022) “High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios” Cell, Volume 185, https://www.cell.com/cell/fulltext/S0092-8674(22)00991-6#%20
10. Roslin N, Li W, Paterson AD, Strug LJ. Quality control analysis of the 1000 Genomes Project Omni2.5 genotypes. bioRxiv. 2016;078600–078600.
11. PopGen CLIはICAのバンドルとしてご利用いただけます。その名称はICAのサブスクリプション地域によって異なります。現在、以下の3地域の利用者にバンドルをご利用いただけます。
a. 米国東部(use1)のエンタイトルバンドル「popgen-cli-release-use1」
b. ロンドン(euw2)のエンタイトルバンドル「popgen-cli-release-euw2」
c. シンガポール(apse1)のエンタイトルバンドル「popgen-cli-release-apse1」