ハイスループットシーケンスは、臨床診療および集団遺伝学(PopGen)研究にますます多く採用され、その課題はテクノロジーへのアクセスを得ることから、生成されたデータを効率的かつ正確に最大限に活用することにシフトしています。Illumina DRAGEN(Dynamic Read Analysis for GENomics)Bio-ITプラットフォームは、その包括的な機能により、小規模および大規模なバリアントを高精度かつ高速でコールすることができるため、1つのプラットフォームで有意なインサイトを効率的に提供できます。
New York Genome Centerが発表した1000 Genomes Project1のカバレッジ範囲の広い全ゲノムシーケンスデータにより、代表的なコホート内でこれらのさまざまなコーラーを状況に応じて解釈することができます。これにより、数多くの多様なサンプルからのバリアントコールを観察できるだけでなく、カバレッジデータが不均一であったり、バリアントコーラーの仮定に反する領域でも絞り込みを行うことができます。
ここでは、1000 Genomes ProjectデータにDRAGENプラットフォームを展開し、以下の作業を行いました。
- 小規模および大規模なバリアントを同定、集約し、公開しました2。
- より大規模なバリアントを調査するためにDRAGENが提供するさまざまなインサイトを例で示しました。
- 潜在的なアーティファクトまたはメンデルの法則の仮定に違反する小規模バリアントにフラグを付けるために、コホートサンプルにDRAGENの高精度とフィルタリング機能を活用しました。フラグ付きバリアントも公開されています3。
I)1000 Genomes Projectデータセットに対して使用されるDRAGENスピードおよびバリアントコーラー
DRAGENプラットフォームは、コホートサンプリングから高精度で小規模および大規模なバリアントをコールするために並列して使用する複数のパイプラインを備えています。表1に、バリアントコールに使用できるDRAGENパイプラインについて説明します。
表1:1000 Genomes Project解析に使用されるDRAGENパイプライン
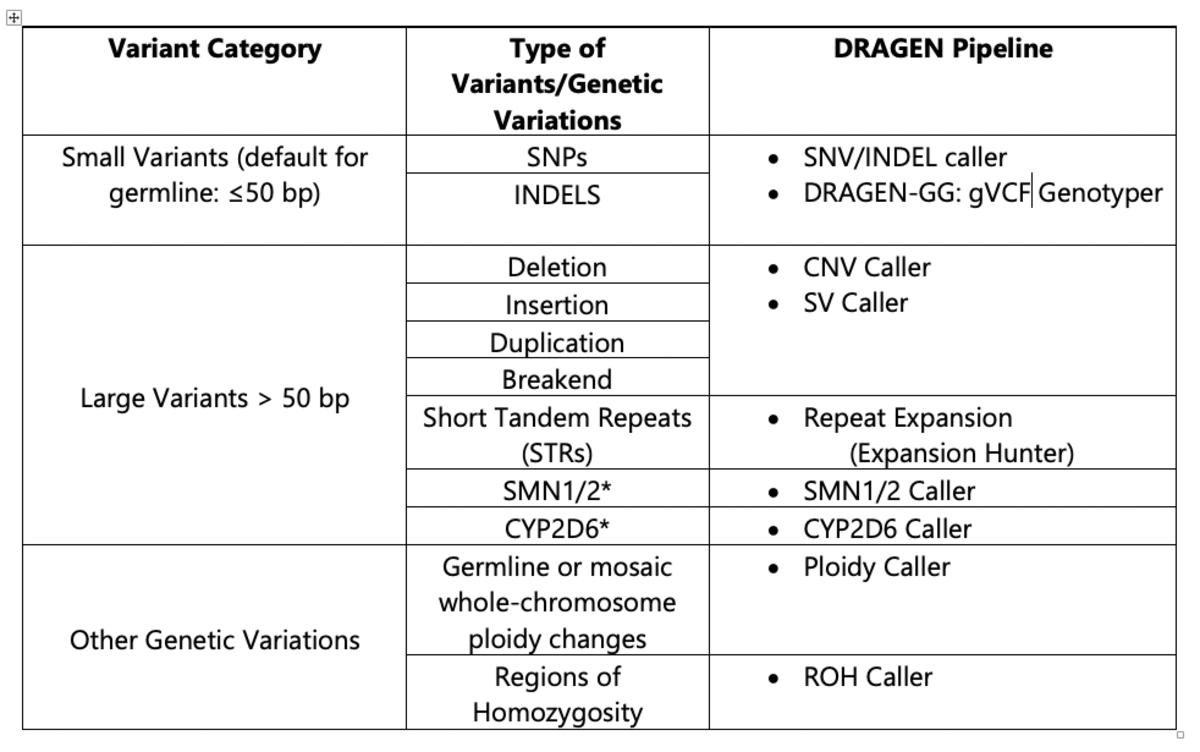
*CYP2D64,5およびSMN 1/26,7パイプラインは、v3.5.7bを使用した1000 Genomes Projectデータセットの現在の解析に含まれていませんが、DRAGEN v.3.7(CYP2D6)およびDRAGEN v.3.8(SMN1/2)では利用可能です。
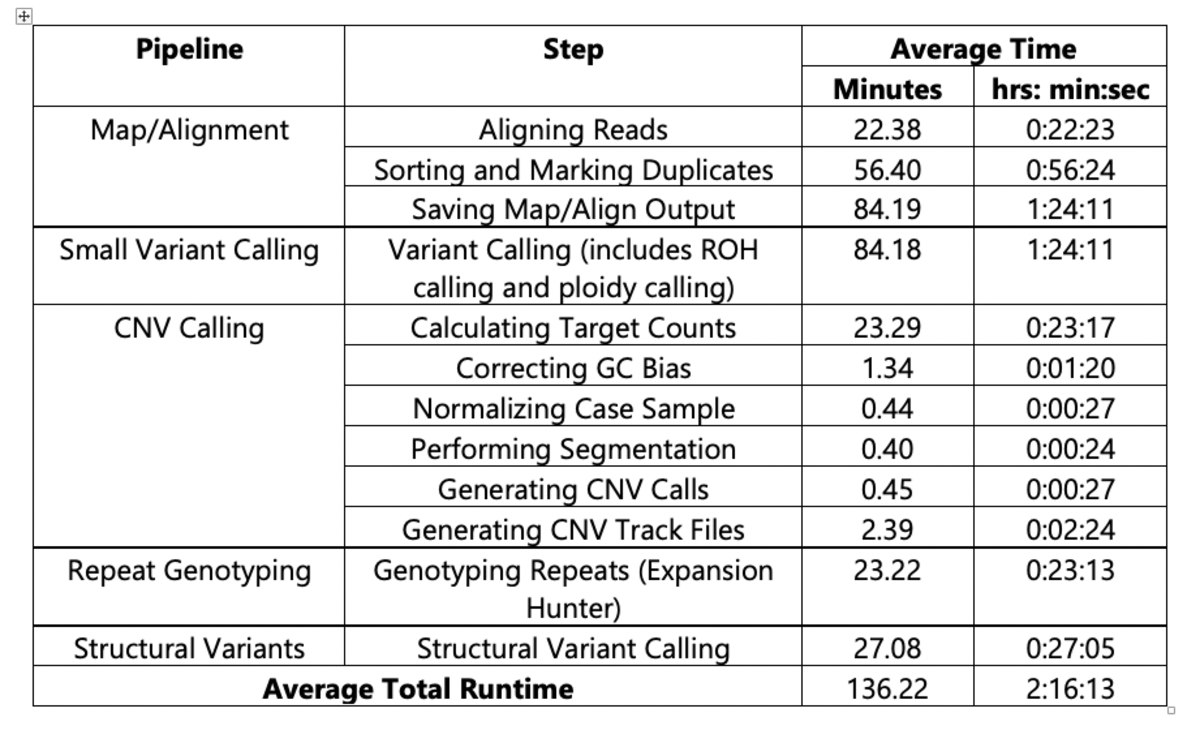
1000 Genomes Projectのデータセット解析では、使用したすべてのパイプラインを単一のDRAGENコマンドでオーケストレーションし、各サンプルに対して1回のエンドツーエンドランで展開しました。DRAGENの高い処理速度の例を示すために、表2および図1では、Illumina Connected Analytics8クラウドコンピューティングプラットフォーム上で、f1.4xlarge AWSインスタンスを使用してDRAGEN v3.5.7bで実行した2504サンプルの解析における平均ランタイムを示しています。
表2:ICAでのDRAGEN v3.5.7bの平均ランタイム(f1.4xlargeインスタンス)
(n=2504、リファレンス:hg38 alt aware)
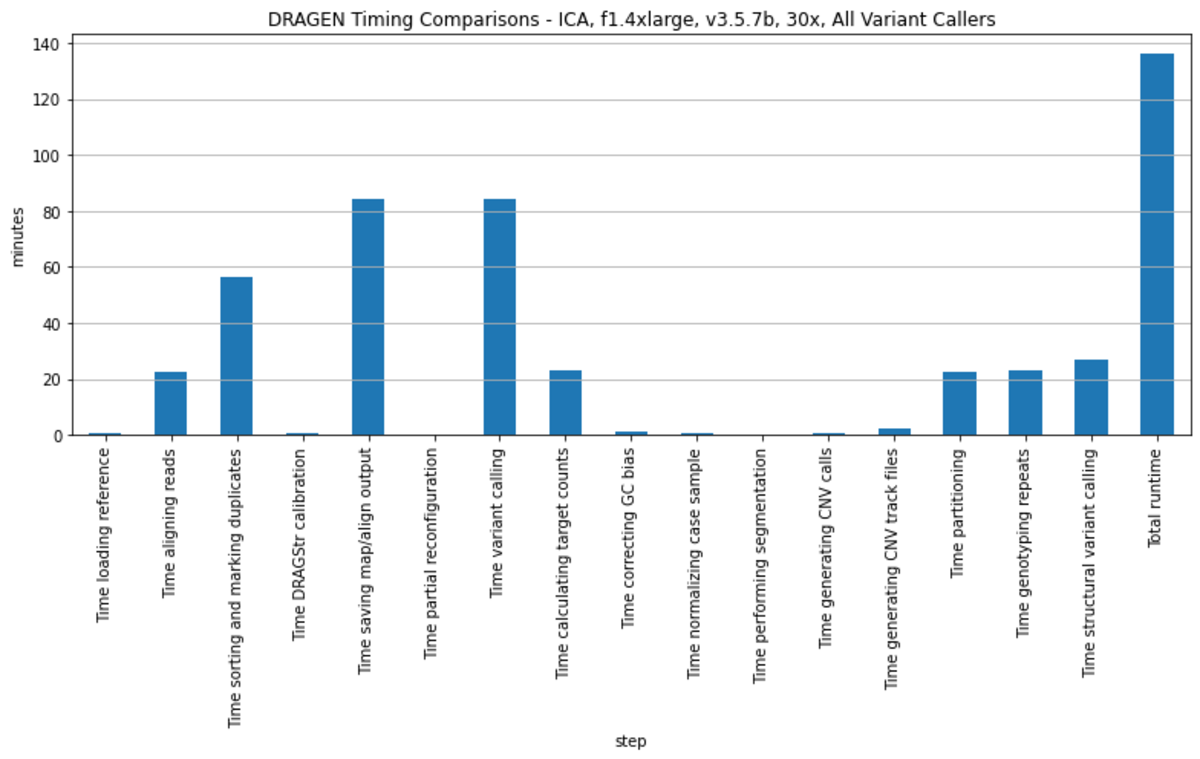
異なるゲノムリファレンス、より大規模なAWSインスタンス(例:f1.16xlarge)、またはオンプレミスのDRAGENサーバーを使用して実行したDRAGENランでは、解析ランタイムを短縮できます。ランタイムの改善は、より新しいバージョンのDRAGENソフトウェアを使用することによっても得られる場合があります9。
II)1000 Genomes Projectデータに対する大規模バリアントコールに使用されるDRAGENパイプラインに関する追加のインサイト
構造多型(SV)は、リファレンスシーケンスと比較して個人のゲノムDNAに見られる大きなゲノムバリエーションです。小さな1塩基変異(SNV)よりも頻度は低いものの、これらの大きな変異は遺伝子の機能を妨げる可能性があり、疾患への影響が実証されています10。
さまざまなタイプの大規模バリアントをすべて同定するために、DRAGENプラットフォームには、個人のゲノム構造のインサイトを収集するために連携して使用する複数のツールが組み込まれています。DRAGENは、リード深度、スプリットリードとペアリード情報、および既知のゲノム遺伝子座位をターゲットとしたカスタムアルゴリズムを用いた補完的なアプローチを採用しています。
本稿では、コピー数バリエーション(CNV)などのSVのサブタイプをコールするために、DRAGEN-SVコーラーをその他のDRAGENパイプラインと組み合わせてどのように使用するかについて説明します。また、DRAGENがコール困難な領域のバリアントをコールするためにターゲットアプローチをどのように展開するかについても説明します(例えば、STRにはExpansion Hunterを使用し、シトクロムP450 2D6酵素の遺伝子コード化におけるバリアントにはCYP2D6コーラーを使用し、脊髄性筋萎縮症に影響する生存運動ニューロン遺伝子のバリアントにはSMN1およびSMN2コーラーを使用します)。
DRAGEN SV Caller
DRAGEN SVコーラーは、オープンソースのManta SVコーラーに由来します11。Mantaと同様に、スプリットリードおよびペアリードの両方のエビデンスを活用して、欠失、挿入、タンデム重複、ブレークエンドなどの構造多型を検出し、スコアリングします。慣例上、構造多型は最小で50bpのサイズまで報告されています。200塩基以上の欠失およびタンデム重複については、SVコーラーはさらに、バリアントとその隣接領域の間の深度変化がバリアントタイプと一致するようにし、これらのバリアントタイプの精度をさらに向上させます。
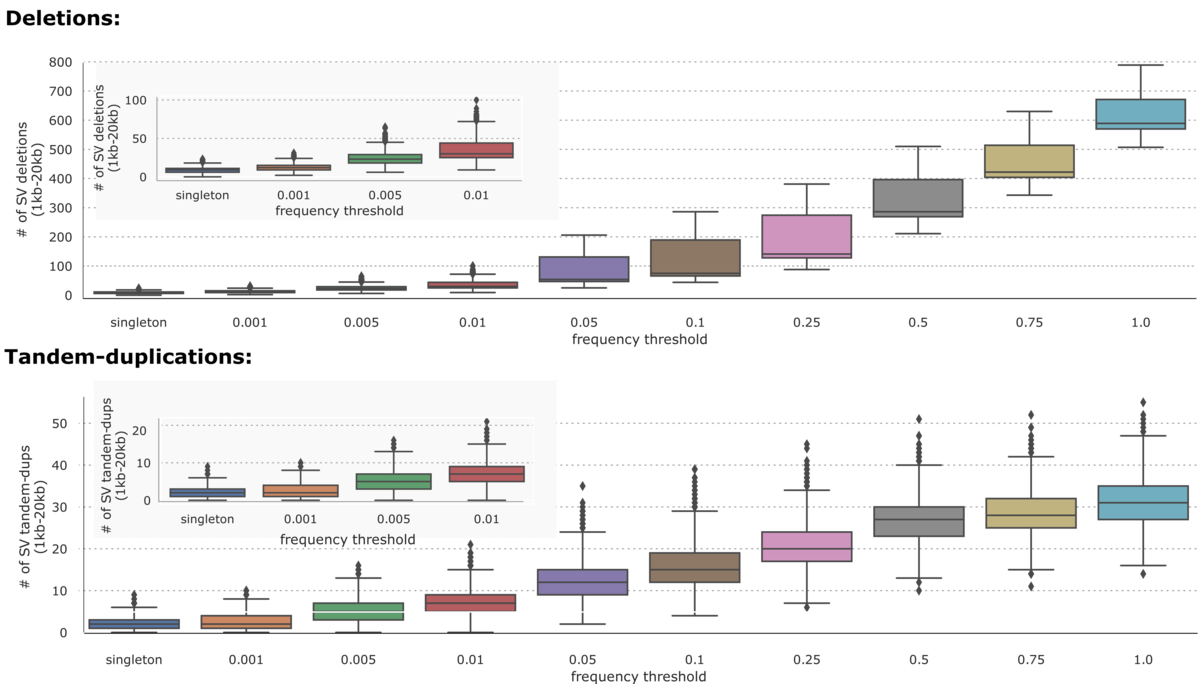
1000 Genomes Projectデータセット(n=2,504)では、DRAGEN-SVパイプラインは、図2に示すように、異なる頻度閾値に分散した36,961件の一意の欠失(1kb~20kb)と7,534件の一意のタンデム重複(1kb~20kb)をコールすることができました。
DRAGEN CNV Caller
DRAGEN CNVコーラーは、深度ベースのアプローチを使用して、隣接する生殖系列コピー数の領域にゲノムをセグメント化します。これは生殖系列由来のバリアントを前提として動作するため、モザイクバリアントの検出には特化していません(ただし、高純度のモザイクCNVはCNVコーラーの出力VCFファイルによって報告される場合があります)。一般的な目安として、このコーラーは30×のゲノムにおいて10kb以上の変異に対して非常に優れた性能を発揮しますが、この性能はカバレッジの深度に比例します。非常に大きなゲノムCNVは、CNVコーラーに認識され、そのソース染色体上の倍数性の変化にも反映される可能性があります。深度ベースのサポートに加えて、さらに、スプリットリードまたはペアリードのサポートがある場合は、DRAGEN-CNVコーラーだけでなく、DRAGEN-SVコーラーにもCNVが表示される場合があります。
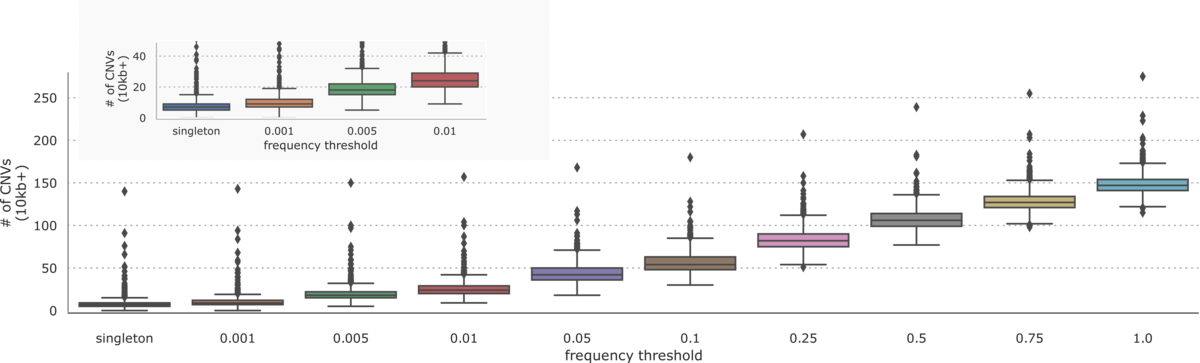
1000 Genomes Projectデータセットでは、DRAGEN CNVパイプラインは、図3に説明される頻度分布で、10kbを超えるサイズの2,891件の一意なCNVをコールすることができました。
サンプル全体のCNVを集約すると、10kbを超えるサイズのコールがサンプルあたり約150件見られます。小規模バリアントと同様に、多くのCNVは非常に一般的であり、1%の頻度閾値で約25の変異が残り、サンプルごとに6つのシングルトン変異が存在することがわかります。
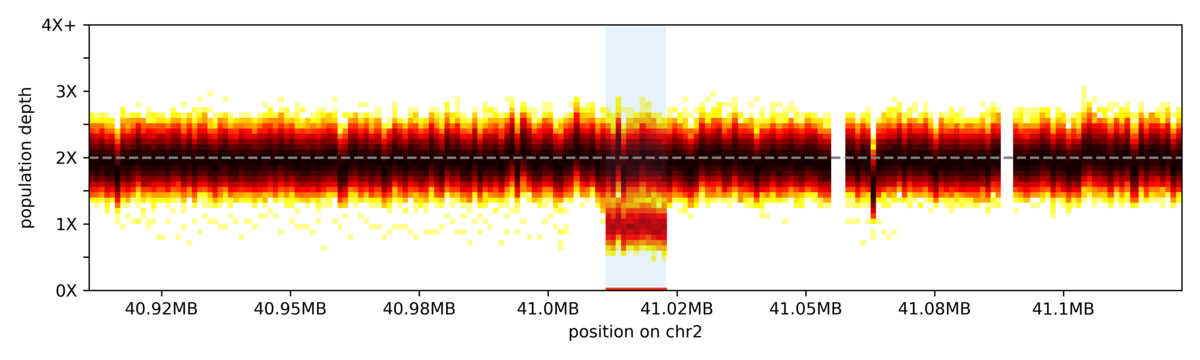
ヒートマップの垂直スライスはカバレッジbinを表し、各スライスの色強度は、その所定のビンの1000ゲノムコホート全体のサンプルカバレッジ深度の分布を表します。強調表示された領域は、一般的な欠失の座標を示します。
DRAGEN ExpansionHunter
STR/リピート伸長は、挿入されたシーケンスが反復モチーフの追加のコピーを構成する、特殊なクラスの挿入バリアントです。SVコールの方法論(挿入されたシーケンスのde-novoアセンブルに依存している)により、これは依然としてコールが難しいバリアントクラスです。それにもかかわらず、DRAGENプラットフォームは、バリアント構造の事前知識を利用して、シーケンスグラフ(ExpansionHunter)を使用してリピート伸長を正確にコールすることができます12,13,14。DRAGENチームは、遺伝学文献から臨床的に意義のあるリピート伸長をキュレーションし、このバリアントコーリングパッケージの一部として既知のリピート伸長の報告を組み込みました。
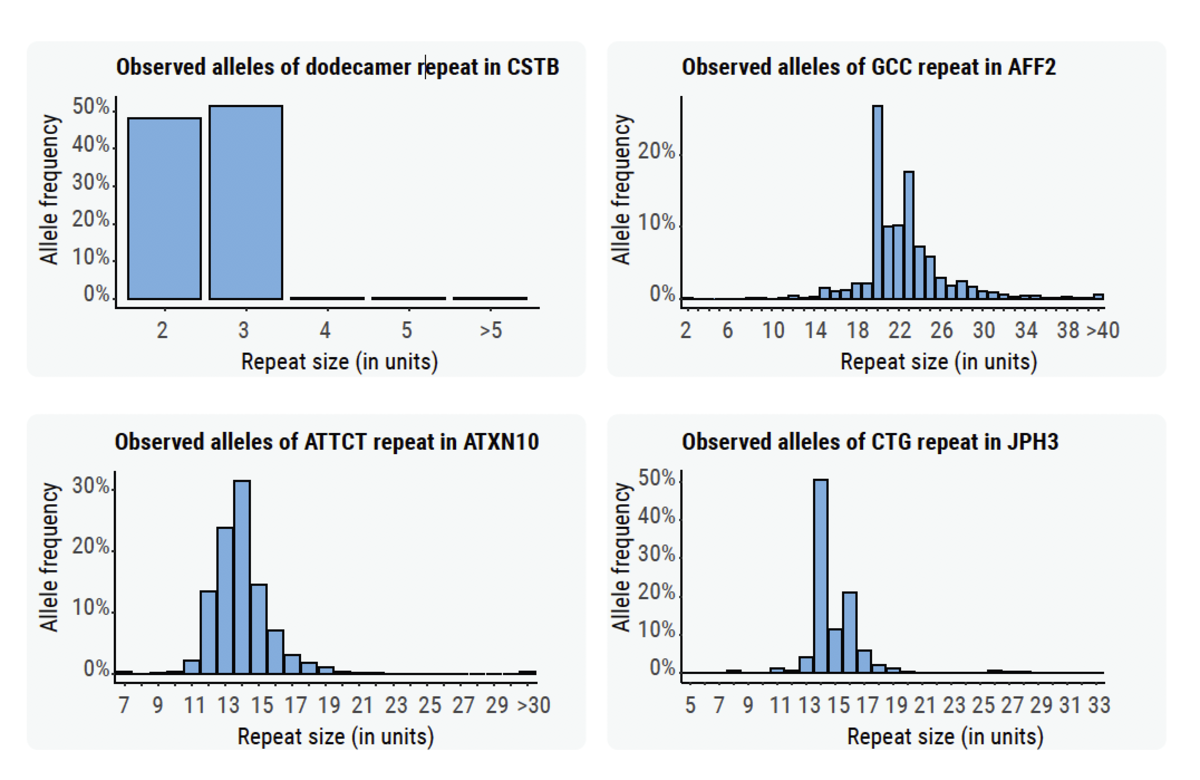
1000 Genome Projectのサンプル全体にわたり、一部のSTRのサイズ分布を生成しました(図5)。
DRAGEN Ploidy Caller
多くの病態において、基準倍数性からの変化が観察され、DRAGENの倍数性コーラーはすべての常染色体および性染色体の平均倍数性レベルを報告します。このコーラーは、生殖系列の全染色体倍数性の変化またはモザイクバリアントを約15%のモザイク純度まで報告することができます。これらの変化はDNAコピー数によって観察され、狭義の構造多型として扱われる場合もあれば、そうでない場合もありますが(多くは構造変化と呼ばれます)、実際的な解釈の観点から、これらはSV(構造多型)としてまとめて議論に含まれています。
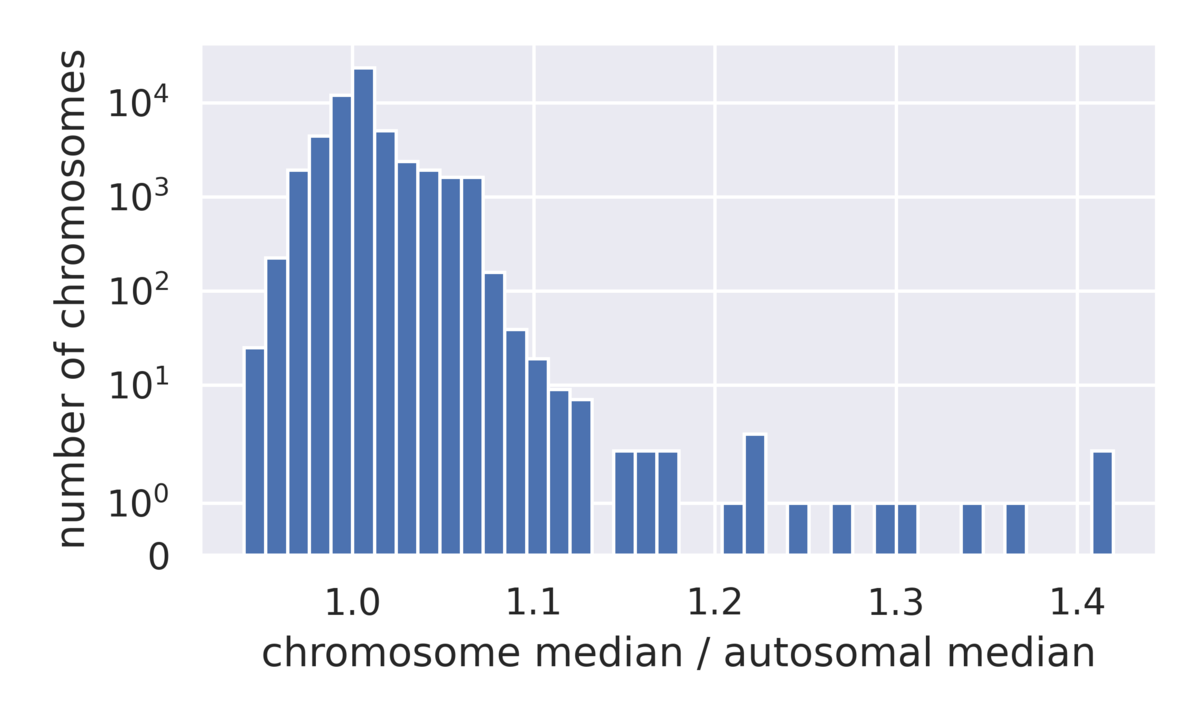
1000 Genomesデータを使用して、多様なサンプルアレイにわたる倍数性コールの分布を調べることができます(図6)。これにより、少数のサンプルで構成されることが多い従来のベンチマーキングデータセットでは実現不可能な方法で、全染色体倍数性の変化の検出限界をより詳しく理解することができます。このサンプルプールにおける染色体カバレッジの評価により、染色体の大部分が予想されるカバレッジの90%~110%であるのに対し、少数の外れ値ではカバレッジが増加していることがわかります。
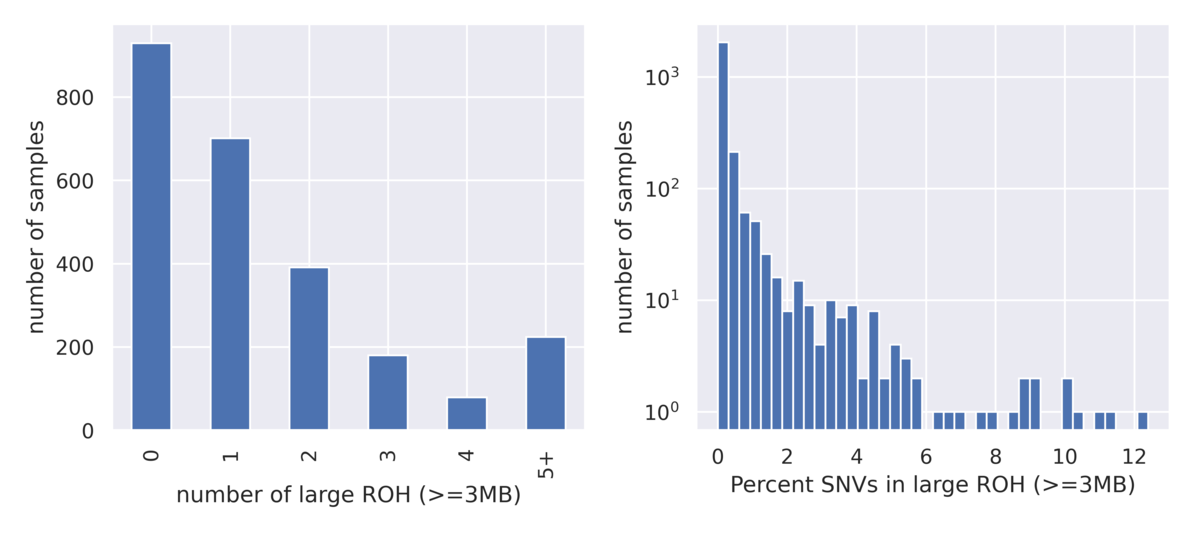
DRAGEN ROH Caller
DRAGENは、大規模なホモ接合性ラン(ROH)コールもサポートしています。このコーラーは小規模バリアントジェノタイプのデータに基づいて動作し、ホモ接合性バリアントがヘテロ接合性バリアントよりも多く現れる領域を強調表示します。大規模なROHコールは、片親性イソダイソミーの存在や、親同士の血縁関係を明示するために使用できます。ROHの自然変異が集団内で異なるため、1000 Genomes Projectデータセットは、典型的な集団におけるROHブロックの基準となる期待値を設定する上で非常に有益であり、臨床事例を異常としてフラグ付けし、さらなる調査のための経験的な閾値を設定するために使用できます(図7)。
III)DRAGENプラットフォームを使用した複数のデータタイプの組み合わせ:不均衡な転座の例
1000 Genomes Projectデータセットの幅広さにより、標準的なベンチマークサンプルでは得られないさまざまな種類の希少なバリエーションを観察することができます。その一例として、NA20533に見られる不均衡な転座があります。これらの大規模なゲノム再構成は極めてまれであり、臨床サンプルで観察されるとほぼ常に病原性を示します。この例では、不均衡な転座は体細胞系のアーティファクトであると考えられます。それでも、臨床的に意義のあるバリアントタイプの例として使用することができます。
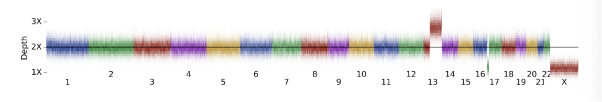
倍数性コーラーの出力を見ると、さらなる調査が必要な異常染色体が見られます。
PLOIDY ESTIMATION,,Autosomal median coverage,36.47
...
PLOIDY ESTIMATION,,13 median / Autosomal median,1.24
...
この例では、DRAGENのtarget.counts.gc-correctedファイルを使用して、ゲノム全体のカバレッジを視覚化します(図8)。これを染色体カバレッジの背景分布と比較すると、これは明確な外れ値であることがわかります。
データはヒートマップとして表示され、画像のスライスは100kbのゲノム間隔での深度binの分布(.target.counts.gc_normalizedファイルから)を示しています。
上の図8では、13番染色体に非常に大きな重複が見られ、17番染色体には大きな末端欠失が見られます。これは不均衡転座を示唆しています。CNVコール(表3)に移ると、1回のLOSSコールで欠失が表示され、重複は5つの別々のコールに分割されます。この分割は、一般的なコピー数バリエーションやノイズが影響して、バリアントコーリングが複数の部分に分かれるためであり、このような大きなCNVではよく見られる現象です。
表3:不均衡転座に隣接するCNVに対応するNA20533用のCNV.VCFファイルのサブセット

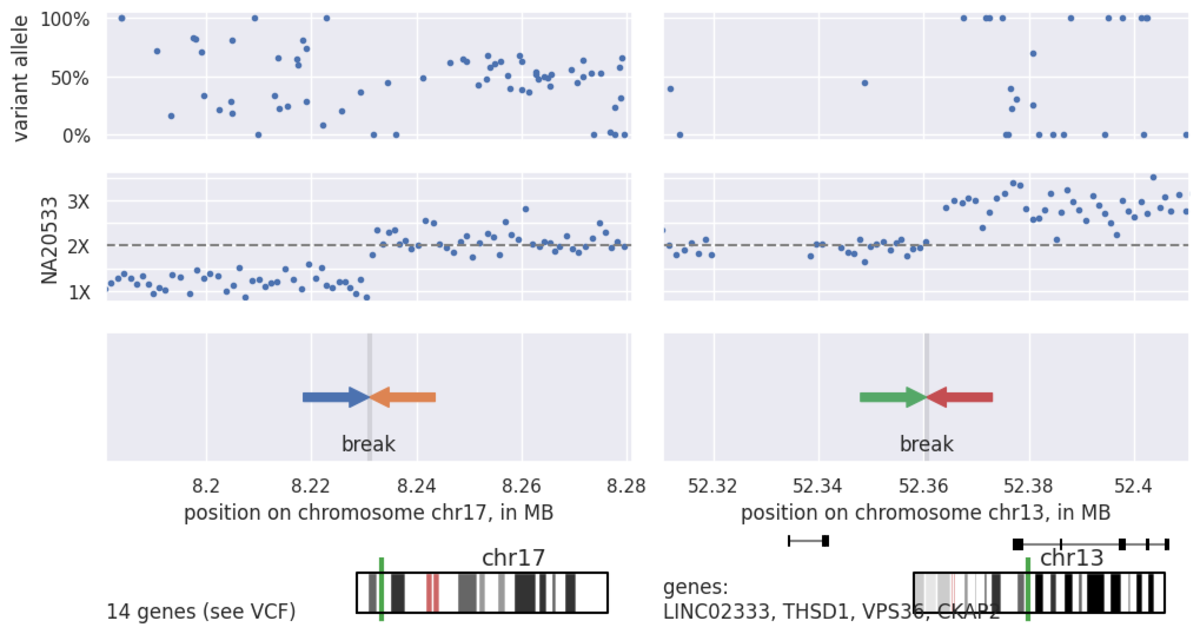
ドットは`.target.counts.gc_normalized`ファイルからの正規化され、bin分割されたカバレッジデータを表します。
このカバレッジデータを見ると、この不均衡な転座のブレークポイントを確実に推定しているという信頼感が得られます。最後に、これらの転座はゲノム再配列とコピー数の変化をもたらすことがわかっているため、CNVで定義されたブレークポイントの近くにある構造多型コールファイル`.sv.vcf.gz`をペアリードデータおよびスプリットリードデータを介して調べ、このバリアントのエビデンスとすることができます(表4)。
表4:NA20533における転座ブレークポイントのVCF記録

この例では、DRAGENパイプラインが、多くのスケールで希少な大規模バリアントの観察を容易にし、リード深度とスプリットリードの両方の観点からゲノムを観察することができることを示しています。CNVコーラーおよび関連するカバレッジデータでは、非常に信頼性の高いコールとバリアントの明確な解釈が得られます。一方、構造多型コーラーでは、ブレークポイントの解決と、この特定のゲノム再構成の形成メカニズムについて理解が得られます。
IV)小規模バリアントコーリングの精度
サンプル間のバリアント表現を統一し、コホート解析を可能にするために、Illumina DRAGEN(Dynamic Read Analysis for GENomics)Bio-ITプラットフォーム、バージョン3.6.3のgVCF Genotyperコンポーネントを使用しました。
gVCF Genotyperは、コホート内の各サンプルから小規模バリアントコーラーの出力を取得し、すべてのコホートメンバーの任意のサンプルに見られるすべてのバリアントをジェノタイピングします。特定のバリアントを持たないサンプルでは、小規模バリアントコーラーの出力に含まれる深度情報を基にホモ接合性リファレンスの信頼度が推定されます。ただし、gVCF Genotyperは、それ以外の方法で集団情報に基づいて遺伝型を調整することはありません。出力は標準マルチサンプルVCR15形式で表示され、下流コホート解析に使用できます。
その出力を、1000 Genomes Projectからの2,504サンプルで構成されるコホートコールセットの形式で、すべてのサンプルとアノテーション付き集団内頻度でバリアントをジェノタイピングした状態でリリースしています。このコールセットの構築に使用されたサンプルごとの小規模バリアントコールは、以前のリリースで利用可能になりました16。
生殖系列の小規模バリアントに広く採用されているGATKベストプラクティスワークフロー17で、独自に生成されたコールセットと比較することで、得られたデータセットの高品質を実証します。
1000 Genomes ProjectデータにおけるDRAGEN PopGenパイプラインによるスケーラブルなコホートのコール
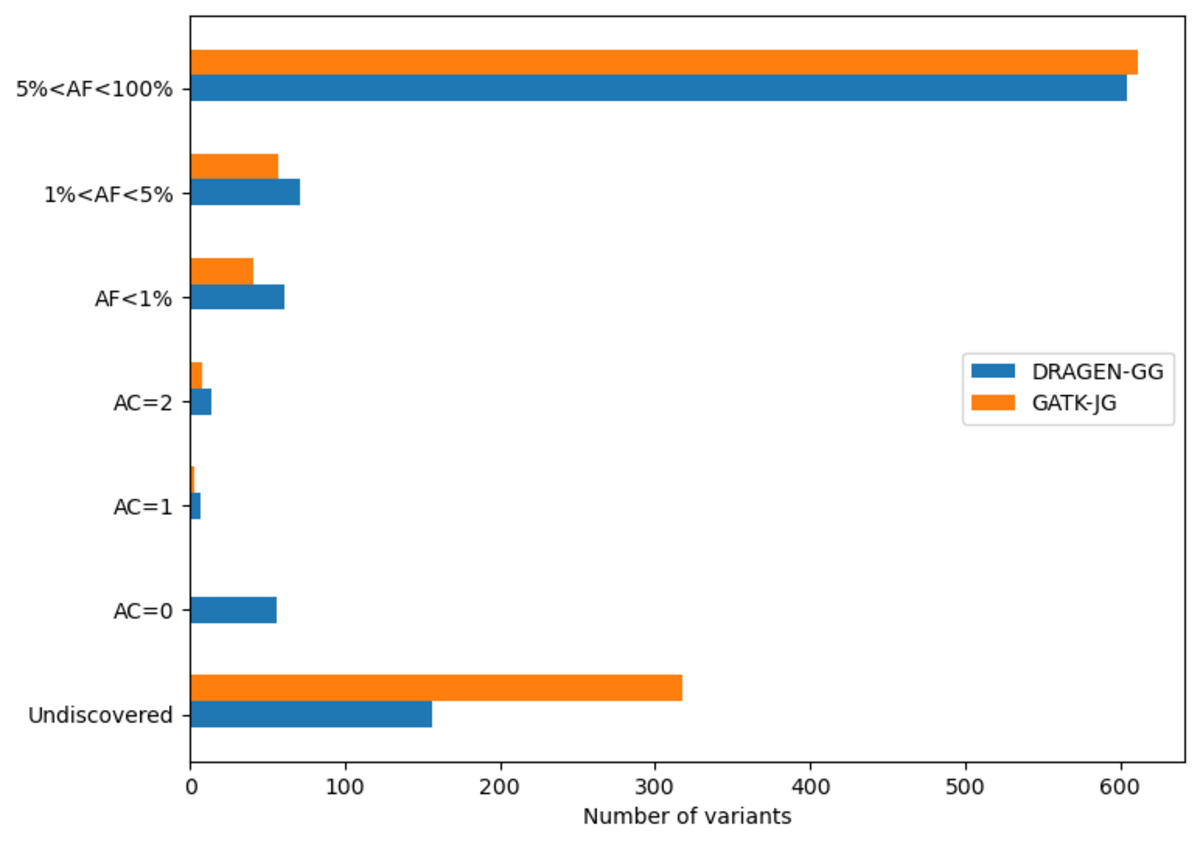
DRAGENコホートのリリースは、各染色体のマルチサンプルVCFで構成され、2,504件の無関係なサンプルが含まれています。全ゲノムにわたって、データセットには合計1億5,100万の座位が含まれ、1億3,800万のSNPと1,800万のIndelがあります。その他のバリアントコーラーとは異なり、DRAGENはエビデンスはあるものの、十分な信頼度でコールされない候補アリルを出力します。そのため、出力された座位の一部はアリルカウント(AC)0となっており、残りの座位の一部にはAC=0のアリルとコールされたアリルの両方が含まれています。これらのアリルの潜在的な価値は図13に示されています。必要に応じて、これらを次のコマンドで削除できます。
bcftools view -a {in.vcf.gz} | bcftools filter -e 'ALT="."' -Oz -o {out.vcf.gz}
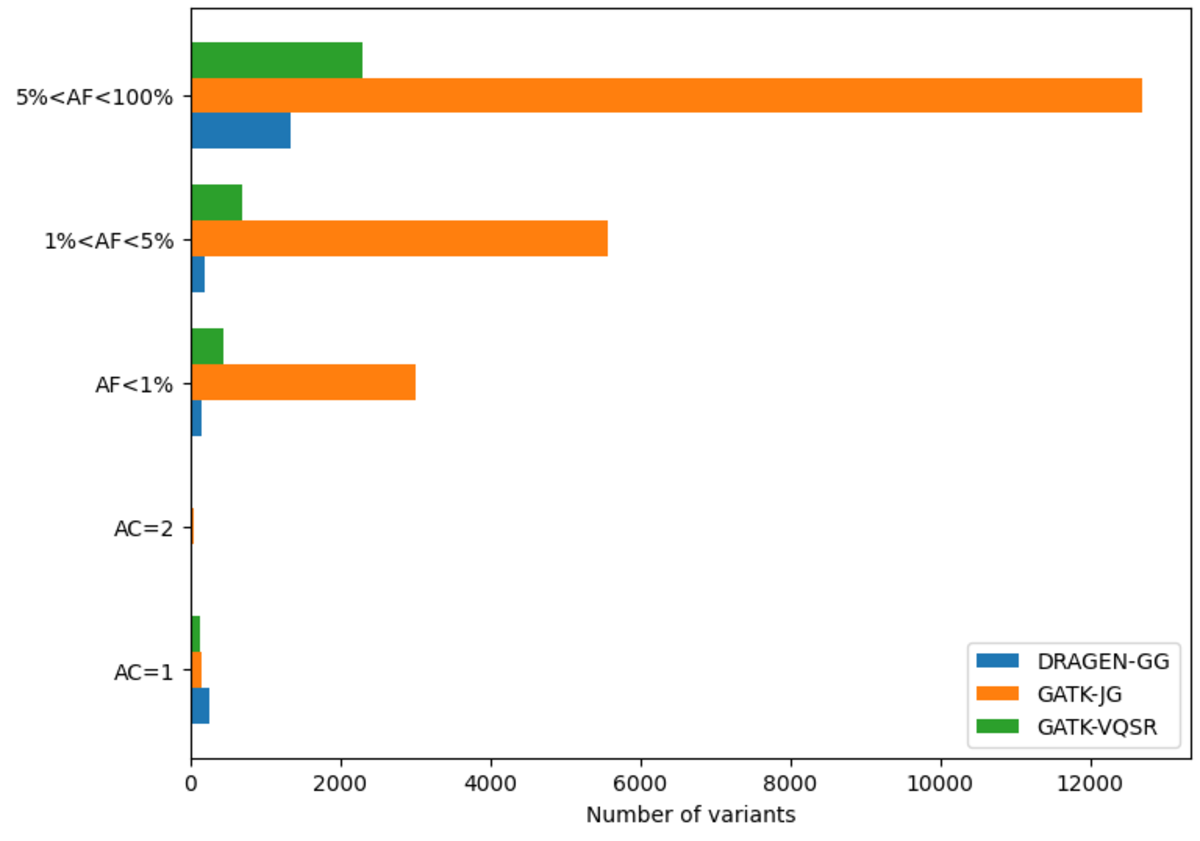
AC=0の記録を除き、1億2,700万の座位があり、1億1,400万のSNP、1,500万のIndel、800万のマルチアリルバリアントがあります。比較すると、類似のGATKコールセットには1億2,000万の座位が含まれ、1億800万のSNP、1,200万のIndel、900万のマルチアリルバリアントがあります。図10は、これらのバリアントのアリル頻度スペクトル全体の分布を示しており、DRAGENが特に稀なバリアントを多くコールしていることが明らかになっています。具体的には、DRAGENの出力には、頻度5%未満のバリアントが1300万件多く含まれています。
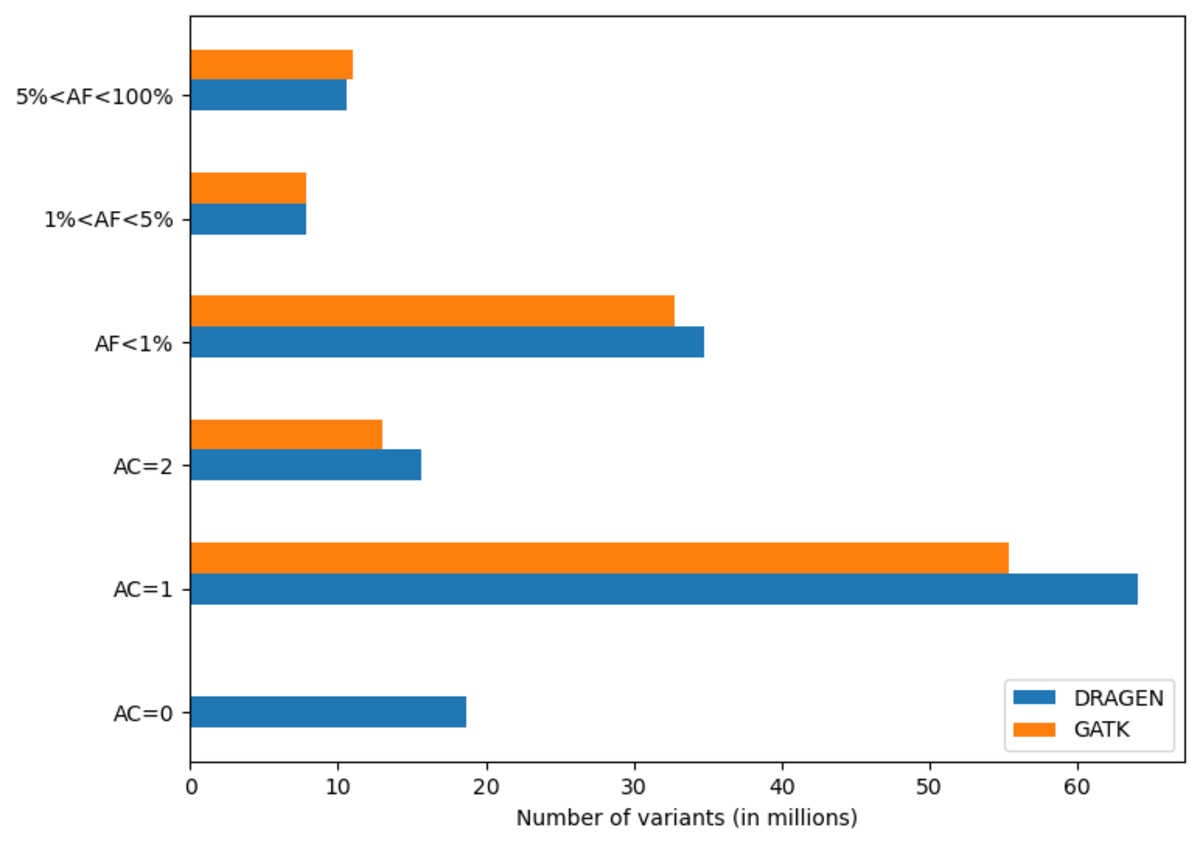
この図では、完全なコールセットが使用されました。GATKでは、VQSRフィルタリング後のバリアントが少なくなります。
DRAGENコホートコールの精度
DRAGENとGATKで得られた集団コールセットの精度を、バリアント真理値セットに対するエラー率、Hardy-Weinberg平衡からの逸脱、トリオにおけるメンデルの法則違反など、さまざまなメトリクスを使用してベンチマークを実施しました。
エラー率
まず、NIST Genome In a Bottle(GIAB18)コンソーシアムが発表した真理値バリアントを含む、十分に特性評価されたサンプルで偽陽性/偽陰性のカウントを計算しました。この正解サンプル(NA12878)は、もともと1000 Genomes Projectコホートの一部として含まれていました。NA12878を表す列をマルチサンプルVCFから抽出し、NISTデータセットのバージョン3.3.2における高信頼度領域を使用して、真理値セットとバリアントを比較しました。図11は、DRAGENコール ("DRAGEN-GG") およびGATK出力からの2つのコールセット、ジョイントジェノタイピング後のすべてのバリアント("GATK-JG")とバリアントクオリティスコアの再キャリブレーションに合格したバリアントのみ ("GATK-VQSR") のエラー数を示しています。DRAGEN-GGは、SNPとIndelの両方において、偽陽性と偽陰性の数が最も少なくなっています。
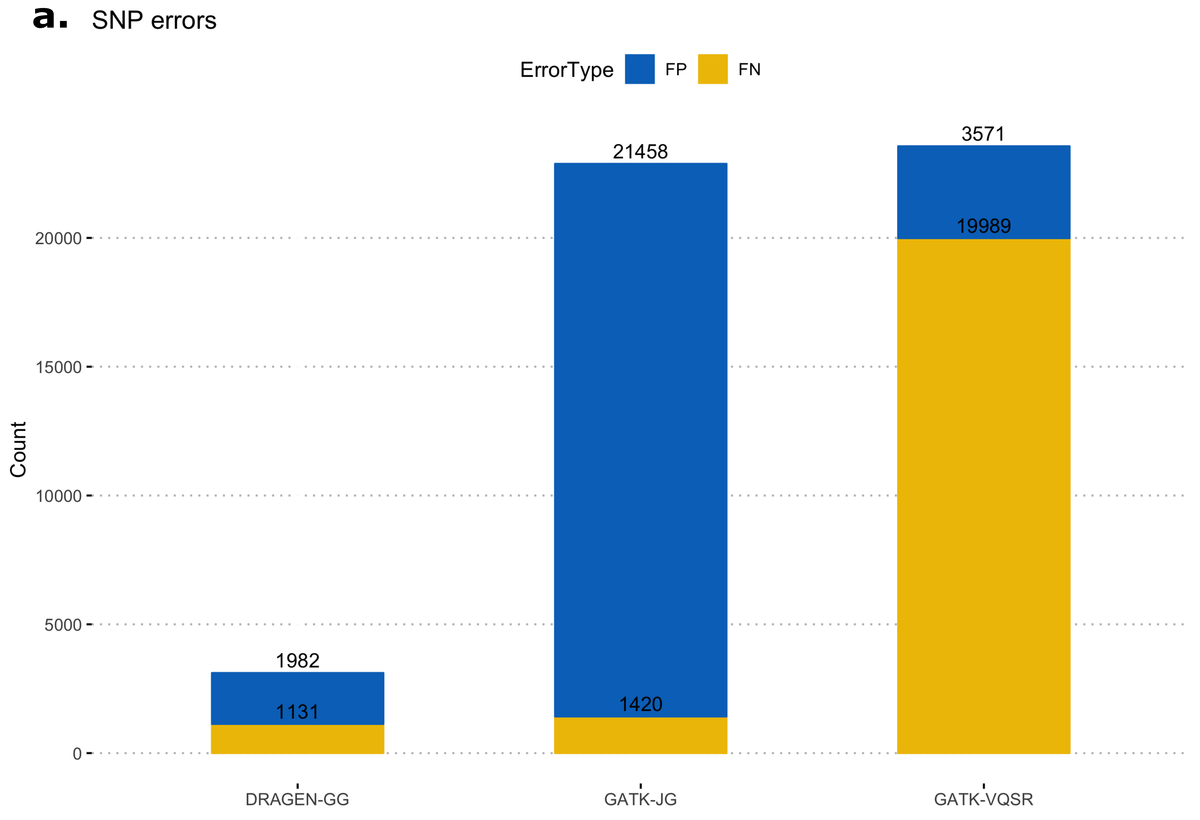
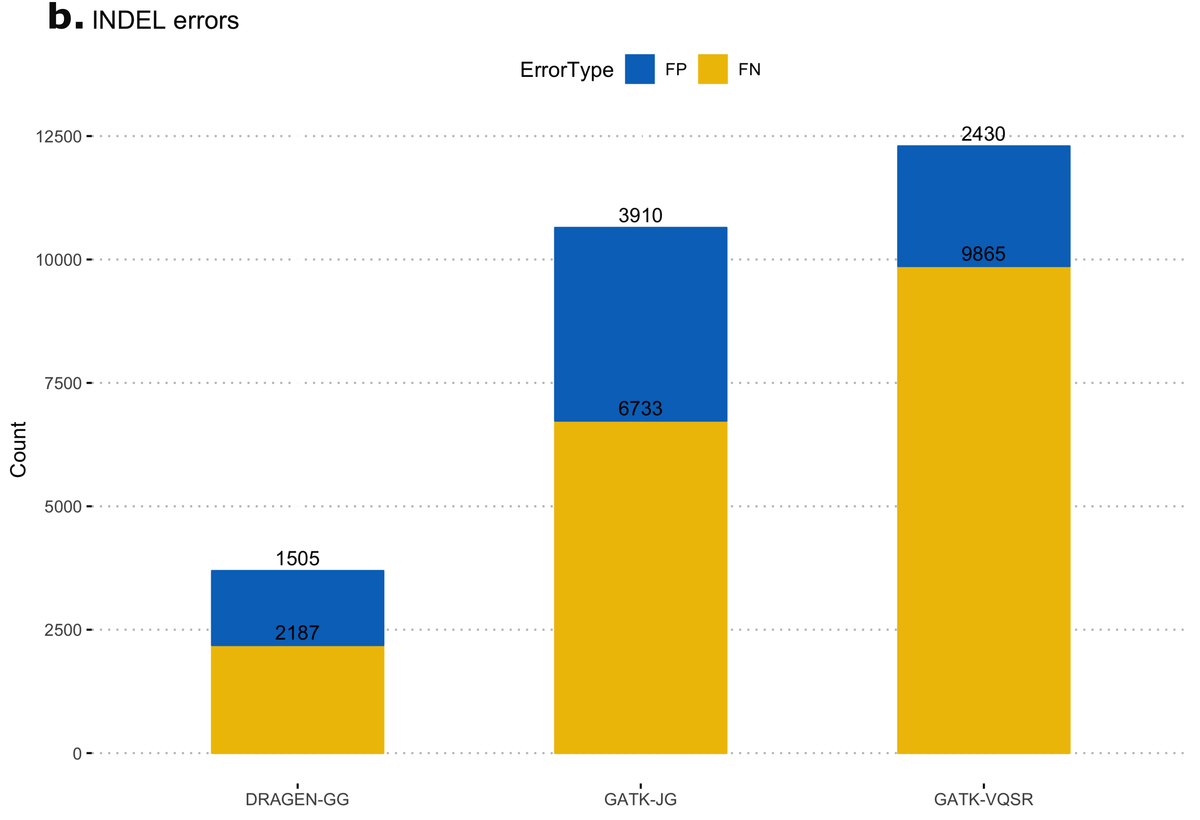
Hardy-Weinberg平衡からの逸脱
次に、Hardy-Weinberg平衡からの逸脱を測定し、各コールセットでアーティファクトの兆候を示す座位の割合を比較しました。GATK VariantAnnotatorを使用して、各座位のExcessHetメトリクス(過剰なヘテロ接合性)を表すHardy-Weinberg平衡検定のPhredスケールp値を計算しました。値が高いほど、技術的なアーティファクトが発生する可能性が高くなります。図14は、DRAGENデータセットがHardy-Weinberg平衡(HWE)を満たしている座位の割合が最も高いことを示しており、DRAGENのバリアントコールの高精度性を強調しています。また、ExcessHet ≥ 28.69(p値はHardy-Weinberg期待値から3標準偏差以上離れている場合に相当)を持つ座位をフィルタリングの対象としてフラグ付けしました。その結果、DRAGENの座位のうちフラグ付けされたのは0.05%未満でした。この情報は、ExcessHetメトリクスを含む座位専用VCFファイルとしてDRAGENコホートコールと併せて提供されており、その他の一般的に計算された座位アノテーションも含まれています。すべての情報を1つのファイルに統合したい場合は、以下のコマンドを使用してDRAGENの出力VCFと座位専用VCFをマージできます。
bcftools annotate –a {sites.vcf.gz} -c INFO,+FILTER –Oz –o {output.vcf.gz} {dragen_calls.vcf.gz}
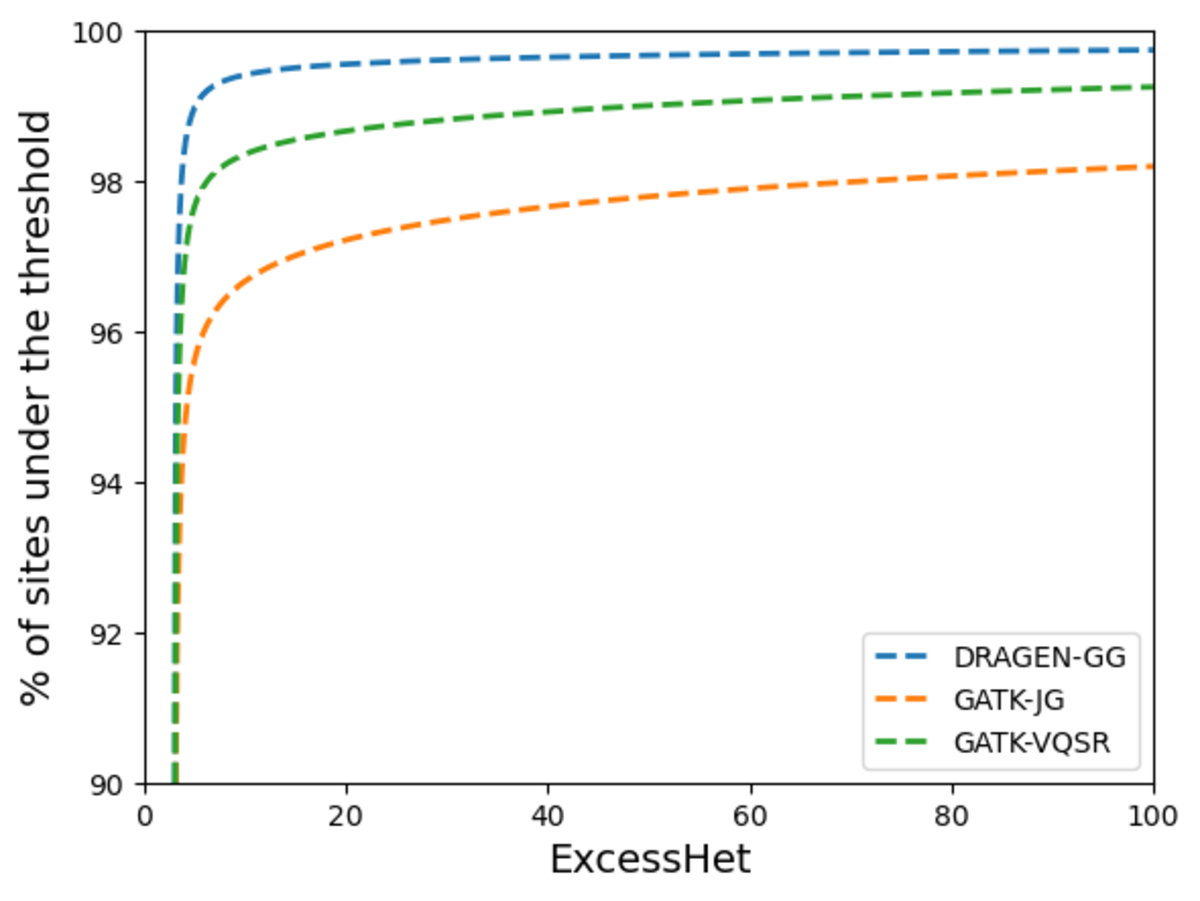
この値が高いほど、サンプルが無関係である場合に技術的アーティファクトが発生する可能性が高くなります。
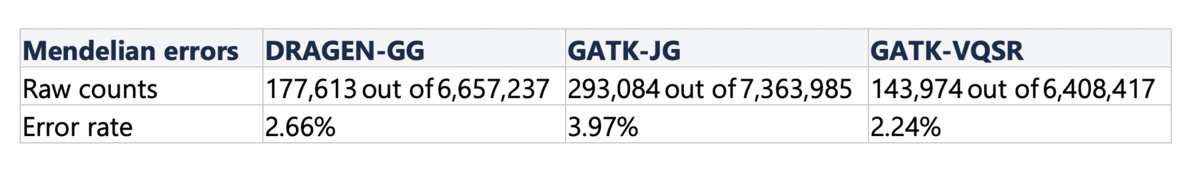
メンデルエラー
最後に、コホートの一部であるサンプルNA20891、NA20882、NA20900で隠れたトリオのメンデルエラー率を計算しました。家族関係の規則への違反は、ゲノムの高信頼度領域内のバリアントに限定されていないため、真理値セットよりも精度をより広く評価するための有用なメトリクスです。表5は、トリオのメンバーの少なくとも1人にバリアントがある座位の総数に対するメンデルエラーの数を示しています。DRAGEN-GGとGATK-VQSRの性能は同等ですが、GATK-VQSRのコール数が全体的に少ないために、わずかに上回っています。
表5:パイプラインあたりのメンデルエラー率
謝辞 Shyamal Mehtalia氏、Egor Dolzhenko氏、Christopher Saunders氏、Heidi Norton氏、およびRami Mehio氏に対し、本稿で示したテストに関与、サポート、レビューし、データにアクセス可能にしてくれたことに感謝を申し上げます。
学術用途向けの詳細情報またはDRAGEN試用版ライセンスについては、dragen-info@illumina.comまでお問い合わせください。
外部リンク
https://github.com/Illumina/manta
https://github.com/Illumina/canvas
https://github.com/Illumina/ExpansionHunter
https://github.com/Illumina/SMNCopyNumberCaller
https://github.com/Illumina/gvcfgenotyper
https://github.com/Illumina/Cyrius
AWS上の1000 Genomes DatasetのDRAGEN再解析
データにアクセスするには、AWS CLIまたはAWS Management Consoleの2つの方法があります。
- AWS CLIでは、ユーザーはaws s3 lsを入力し、以下のリンクをコピー&ペーストする必要があります。
- AWS Management Consoleでは、コンソールにサインインし、https://s3.console.aws.amazon.com/s3/buckets/1000genomes-dragenに進んでください。
データセットリンク
小規模バリアント2504サンプル:
小規模バリアントNA12878の結果:
CNV/SV 2504サンプル頻度:
これらのデータセットについては、このAWSの記事で説明しています。
注釈
- Marta Byrska-Bishop; et al. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. bioRxiv 2021.02.06.430068
- Small variants 2504 samples.
- Small variants NA12878 result.
- Chen, X., Shen, F., Gonzaludo, N. et al. . Cyrius: accurate CYP2D6 genotyping using whole-genome sequencing data. Pharmacogenomics J (2021).
- Cyrius Article
- Chen, X., Sanchis-Juan, A., French, C.E. et al. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet Med 22, 945–953 (2020).
- SMA Article
- Illumina Connected Analytics
- Dragen reanalysis of the 1000 genomes dataset now available on the registry of open data. Figure 3
- Medhat Mahmoud et al. Structural variant calling : the long and the short of it. Genome Biology (2019)20:246 doi: 10.1186/s13059-019-1828-7
- Xiaoyu Chen et al. Manta: rapid detection of structural variants and Indels for germline and cancer sequencing applications. Bioinformatics. 2016 Apr 15;32(8):1220-2. Doi: 10.1093/bioinformatics/btv710. Epub 2015 Dec 8. PMID: 26647377.
- Egor Dolzhenko et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res, 2017 Nov; 27(11):1895-1903. doi: 10.1101/gr.225672.117.Epub 2017 Sep 8.
- Egor Dolzhenko et al. ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics, Volume 35, Issue 22, 15 November 2019, Pages 4754–4756. doi: 10.1093/bioinformatics/btz431
- Reviewer/Expansion Hunter Article
- The Variant Call Format (VCF) Version 4.2 Specification
- Dragen reanalysis of the 1000 genomes dataset now available on the registry of open data.
- Data Collections 1000 Genomes
- Genome in a Bottle