はじめに
体細胞Indel(挿入または欠失)変異は、がんゲノムに多く見られます。The Cancer Genome Atlasの大規模解析では、最も一般的な10の腫瘍タイプ(肺、乳房、脳、結腸など)について約4,000の症例で約8,300の特異的体細胞Indelが明らかになりました。1 DNA損傷修復またはがん遺伝子パスウェイの活性化のいずれかに関与するBRCA1、BRCA2、およびEGFRエクソン20などの遺伝子のIndelについては十分に記録されており、治療介入のバイオマーカーとして機能します。
バイオインフォマティクスツールは、これらのシグナルを精確に抽出する上で重要な役割を果たします。Indelバリアントを精確に同定するため、これらのツールを厳格に評価し、最適化する必要があります。この目的のため、米国食品医薬品局(FDA)は、NCTR腫瘍パネルのシーケンスデータからのIndelコールチャレンジ2を立ち上げ、がんパネルシーケンスデータセットに関する体細胞Indelコールアルゴリズムを開発、検証、ベンチマークする機会をゲノミクス分野にもたらしました。
イルミナは、DRAGEN 4.0体細胞小バリアントコーラー3を携えてこのチャレンジに参加し、DRAGENは、適応性チャレンジ(パネルX)で最高の精度と全面的正確性でIndelコールを生成しました。これにより、体細胞小変異の検出におけるDRAGENアルゴリズムの精度が検証されました。この度の勝利は、前回のPrecisionFDA Truth Challenge V24のマッピング困難領域での生殖系列の小規模バリアントコールにおけるDRAGENの勝利を足がかりとしたものです。
この記事では、体細胞Indelチャレンジについて簡単に説明し、結果を要約して高精度のバリアントコールに寄与したDRAGEN体細胞Indel手法の特徴を説明します。
PrecisionFDAチャレンジの概要
パネルベースのシーケンスの採用が拡大し続けている腫瘍学臨床ラボや翻訳ラボでは、シーケンスを行うコミュニティーが高品質のバリアントコールツールや定番のデータセットにアクセスできることが不可欠です。5FDAは、腫瘍パネルシーケンスを含む次世代シーケンサーによるデータを使用するための標準解析プロトコールや品質管理メトリクスの開発を目的としたシーケンス品質管理フェーズ2プロジェクト6を開始しました。
このチャレンジの腫瘍パネルデータは、10種のUniversal Human Reference RNA(UHRR)がん細胞株の等量混合から作成された人工腫瘍サンプルを使用して、PrecisionFDAチームが作成しました。7基準値セットのIndelは、各細胞株への全エクソームシーケンス(WES)ランを3回行うことにより決定されました。Indelの検証用データセットは、チャレンジ主催者によって各細胞株データセットを使用して、手動でキュレーションされました。この検証用データ(参加者には共有されていない)は、さまざまなパイプラインの提出物を評価するために使用されました。
このチャレンジは2つのフェーズで構成されていました(図1)。フェーズ1では、パネルA(≈3.5 Mb)およびパネルB(≈1 Mb)として知られる2つの腫瘍パネルを用いて、UHRR混合DNAから生のシーケンスデータ(FASTQ)が参加者に提供されました。各ライブラリーは3つの異なるラボで調製され、パネルあたり合計12のシーケンスレプリケートを達成するために4回のシーケンスが行われました。参加者はこれらのデータをそれぞれのパイプラインで処理してデータセットタイプを最適化し、明確に定義されたIndelコールのパイプラインを確立するよう求められました。フェーズ1の終了時に、参加者はパネルAとパネルBの両方について12のバリアントコールの結果を提出するよう求められました。
チャレンジのフェーズ1で結果を提出した研究者らは、フェーズ2に招待されました。このフェーズではパイプラインの変更はできません。各凍結パイプラインの一般化可能性を評価するために、新しいデータセット(パネルX)が参加者と共有されました。パネルXは、Indelコールパイプラインの実世界での性能をシミュレーションすることを目的としていました。多くの場合、臨床ラボや翻訳ラボでは、バイオインフォマティクスパイプラインは最適化され、パラメーターはロックされてその性能が検証されます。その後、ロックされた状態で多くのサンプルにランが実行されます。このチャレンジでは両フェーズにおいて、主催者が共有するターゲット領域の50 bp未満のIndelにのみに焦点が当てられました。チャレンジの詳細については、こちらのウェブサイトをご覧ください。
PrecisionFDAチャレンジの結果
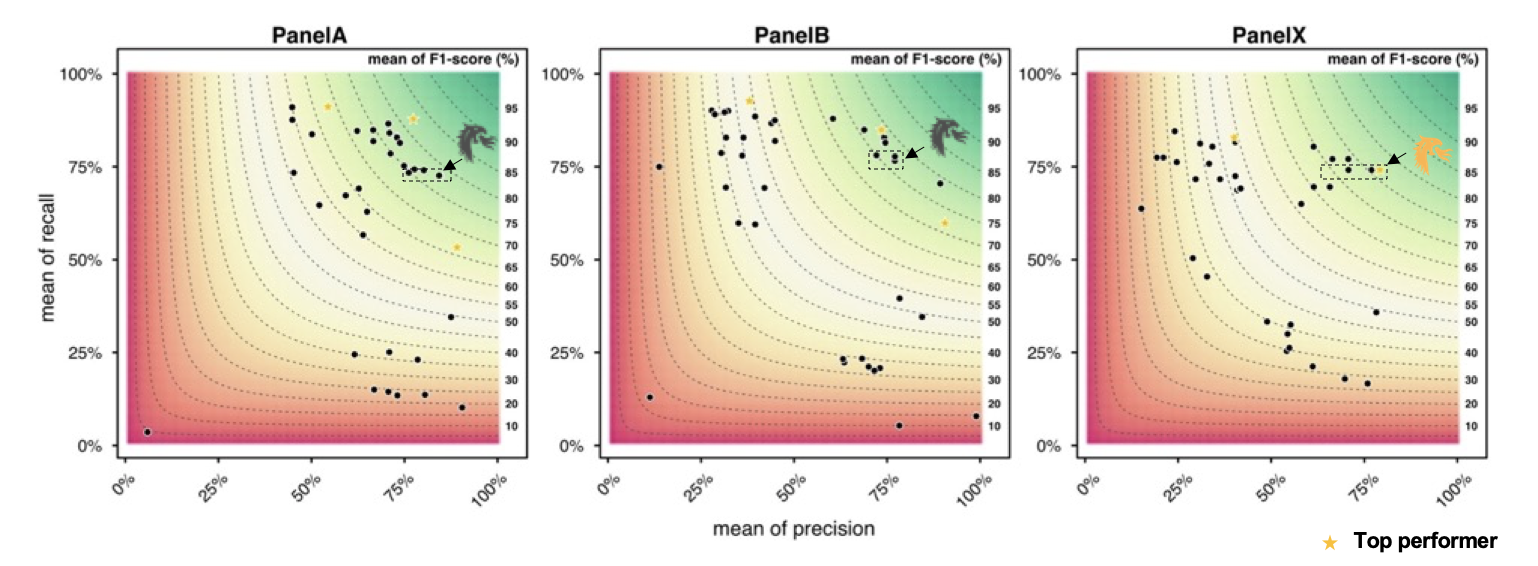
コンペ受賞者の選考基準は、チャレンジが完了した時点で発表されました。各パネルで最高のF1スコア、最高の精度、最高のコール率を達成した最優秀者が発表されました。コール率に有利な精度の過剰な最適化を防ぐため、そのパネルの全提出者の中央値を超えるF1スコアは、プラスの加点をする必要がありました。また、その逆も同様とされました。
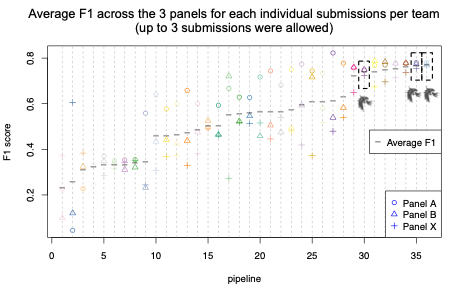
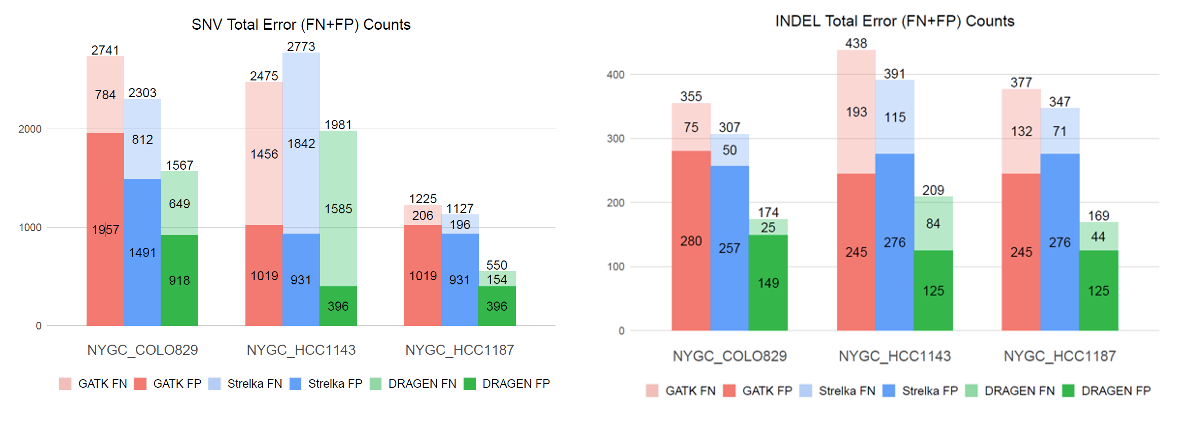
信頼度の高いバイオインフォマティクスパイプラインには、少数の偽陰性(FN)と偽陽性(FP)が必要です。一般的なバイオインフォマティクスパイプラインでは、異なるデータセットにわたって一貫して精確である(すなわち、一般化が可能でなければならない)ことが有益となります。DRAGENは、3つのパネルのすべてにおいて高精度、コール率、F1スコアを達成し、適応性チャレンジのその他の全参加者(パネルX)と比較して最高の精度とF1を達成しました(図2~4)。さらに、DRAGENはすべてのパネルを平均化した場合も最高の精度を示し、性能はパネル全体で一貫していました。これは、DRAGENの高性能が、単一のパラメーターセットを用いて複数の異なるパネルで一般化が可能であることを際立たせており、その他のバイオインフォマティクスソリューションでは頻繁に必要となるパネルごとに固有の最適化を行う必要がないことを意味しています(図2~4)。

DRAGEN体細胞ワークフローとその手法
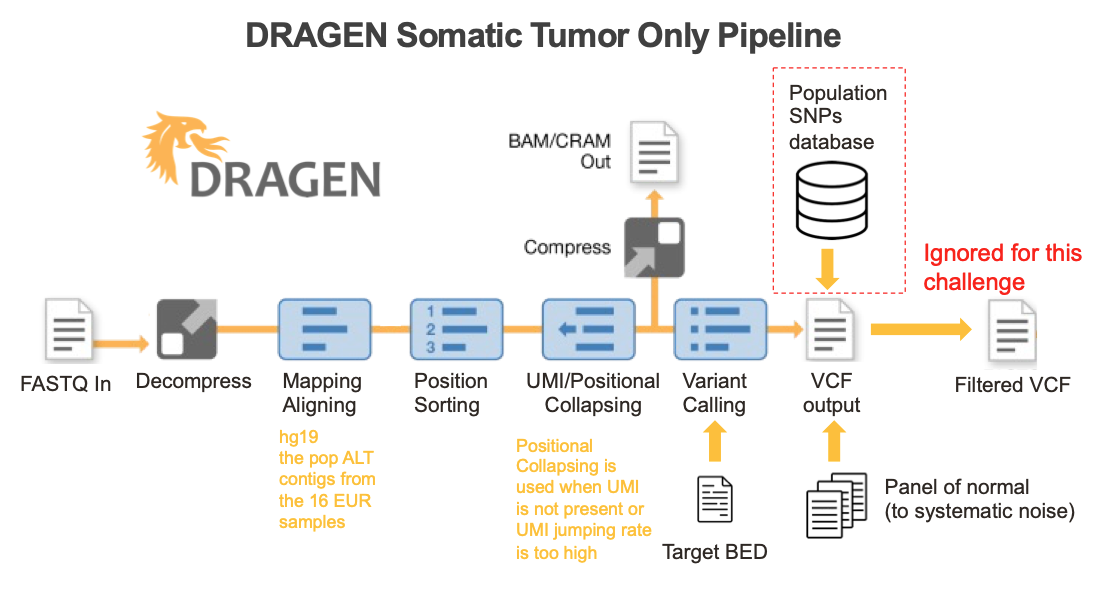
DRAGENは、あらゆる腫瘍パネルから生成された生のシーケンスデータにシームレスなエンドツーエンド処理を行うことを可能にします
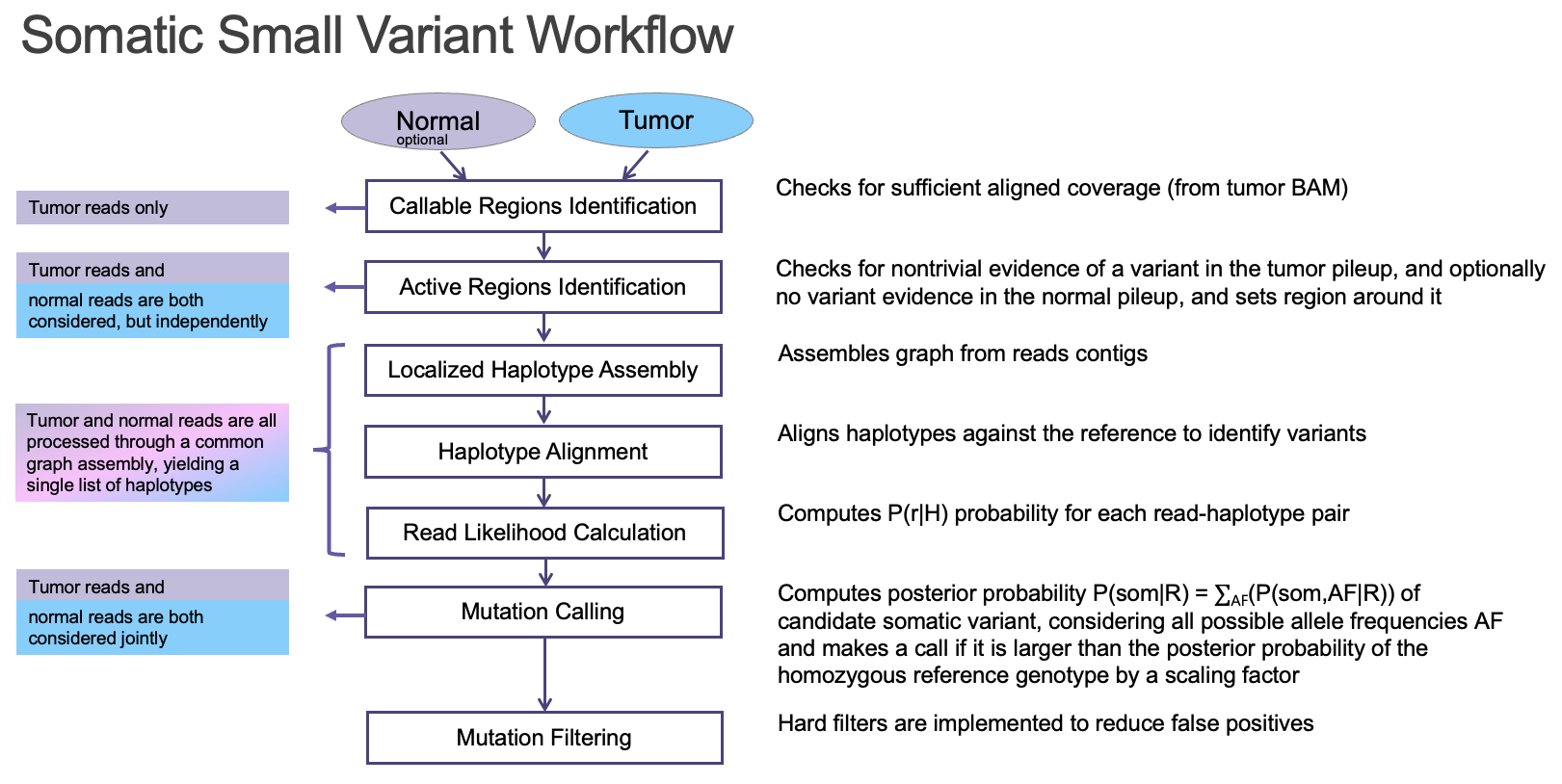
理想的なバイオインフォマティクスパイプラインによって、あらゆるシーケンスデータタイプのシームレスなエンドツーエンド処理が可能になります。この目標を達成するため、DRAGENは未処理のシーケンスリードをリファレンスゲノムにアライメントし、PCRデュプリケートを同定し、固有の分子識別子(UMI)の存在下または不在下でシーケンスエラーを抑制し、体細胞Indelを検出し、低信頼度のバリアントをフィルターしてFPを低減するために活用できる体細胞のクオリティスコアを提供する完成されたソリューションを提供します(図5)。
DRAGEN体細胞小バリアントコールパイプラインに実装された主な機能は次のとおりです。1)DRAGENは、マルチゲノム(グラフ)リファレンスに対する改善されたアライメントを活用しています。これは、ゲノムのマッピング困難領域で高い精度を達成するためには不可欠です。2)DRAGENは、UMIシーケンスの有無にかかわらず、腫瘍パネルデータの処理を容易にします。UMIシーケンスがある場合は、UMIをノイズ補正に効果的に使用する一方で、UMIバーコードが使用されていない場合は、オプションの代替ノイズ低減方法を提供します。3)DRAGENは単一の(高精度の)バリアントコーラーを使用します。複数の異なるオープンソースソリューションを組み込んだ、計算コストが高く、管理が難しいアンサンブルベースのコール法を使用する必要はありません。アライナーやバリアントコーラーに使用される複数のコアアルゴリズムへのフィールドプログラマブルゲートアレイ(FPGA)ベースのハードウェアのアクセラレーションと組み合わせることで、あらゆるシーケンスデータを最高の精度で処理できる最速のパイプラインが得られます。
DRAGENマルチゲノムベーズアライナーによる高精度なIndel検出が可能
DRAGENの主な特徴は、マルチゲノムベースのアライナーであることです。イルミナのDRAGENチーム4やその他8が以前に示したように、マルチゲノムリファレンスを活用することで、マッピング困難領域のリードの曖昧なマッピングが解決します。これにより、アライメントエラーを減らし、バリアントコールの精度を向上させることができます。DRAGENマルチゲノムリファレンスには、集団一塩基多型(SNP)と何百もの代替集団ハプロタイプが含まれています。そのため、リードマッパーに代替ハプロタイプパスに関する情報を提供することで、曖昧にマッピングされたリードを効率的に復元することができます。マルチゲノムリファレンスの構築を促進するため、DRAGEN 4.0+にはマルチゲノム構築ツールキットが同梱されており、研究者は関心のあるマルチゲノムリファレンスをシームレスに生成できるようになりました。DRAGENを用いて行われたこのチャレンジでは、DRAGENマルチゲノムアライナーが使用されました。
DRAGENの柔軟なアーキテクチャーにより、ポジショナルコラプシングを用いるUMIタグの有無にかかわらず効果的なシーケンスノイズ抑制が可能に
UMIは、固有のDNA分子またはRNA分子をPCRデュプリケートと区別するために、1つのリード(シンプレックス)の末端またはペアエンドリード(デュプレックス)の両端に導入される一組の「バーコード」です(図6A)。この確立されたアプローチは、シーケンスノイズを低減するために広く採用されており、シーケンスアーティファクトからの低アレル頻度(AF)変異の識別を促進します。UMIがコラプシングされると、高品質のコンセンサスリードが作成され、特にライブラリーのシーケンスが深い(2000回以上)場合、エラー率が低下し、バリアントコールの精度が向上します。
あるいは、UMIバーコードを使用しない場合は、DRAGENパイプラインが腫瘍パネルのシーケンスデータを処理することができます。この場合、DRAGENはポジショナルコラプシングのアルゴリズムを採用します。つまり、リードがリファレンスゲノムにマッピングされると、アライメント情報と、同位置で開始および終了するリードのシーケンスのみに基づいてリードファミリーが生成されます(図6A)。この場合、そのファミリーのリードの大部分がサポートするバリアントのみが真とみなされ、残りは無視されます。これは、PCRデュプリケートを排除するために従来使用されてきた手法とは対照的です。つまり、後者は同じ開始位置と終了位置にマッピングされたノイズレスリードとノイズの多いリードを区別できず、エラー率が増加することが多く起こります。
1.94 Mbの腫瘍パネル(TruSight Oncology 500)で処理された内製サンプルのシーケンスエラーの比較では、従来のデュプリケート排除法(デュプリケート法とも呼ばれる)と比較して、ポジショナルコラプシングまたはUMIコラプシングを適用した場合のエラー率が半分以下に低下しています(図6B)。さらに、UMIとポジショナルコラプシングは同等のエラー率の低下をもたらすことが確認されています。(UMIを無視して)ポジショナルコラプシングとデュプレックスUMIコラプシングを比較した場合、非常に一致率の高い小バリアントコールが認められました(図6C)。
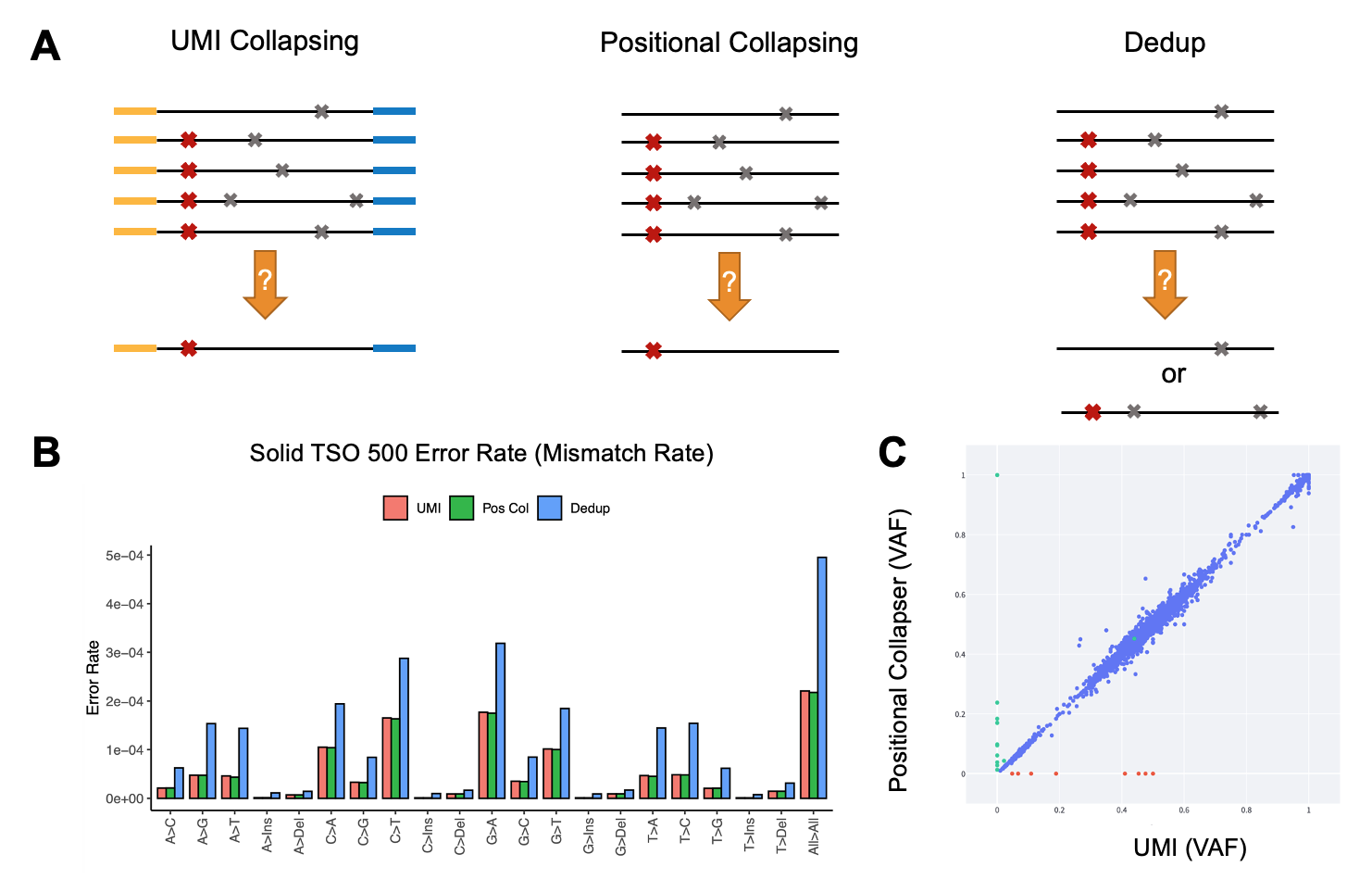
図6A UMIコラプシング、ポジショナルコラプシング、PCRデュプリケート排除の比較。左のパネルでは、リードの黄色と青色の領域はデュプレックスUMIシーケンスを表しています。赤色と灰色の十字記号は、真の変異対シーケンスアーティファクトを表しています。
図6B 3つのノイズ抑制法のエラー率(リファレンスと比較したミスマッチ率)。TSO 500腫瘍パネルでシーケンスされたサンプルを使用して社内で生成されたデータ。
図6CUMIコラプシングとポジショナルコラプシングによって生成されたコンセンサスリードを使用したDRAGENバリアントコール。データポイントは検出された変異のAFを表します。青の点は一致したコールを表し、赤と緑の点はUMIコラプシングとポジショナルコラプシングのそれぞれにのみ存在する変異株を示します。
DRAGENはハプロタイプコーラーの改善から恩恵を受け、低AF体細胞Indelの同定において定評あるその他のツールと比較しても精度が優れています
DRAGEN体細胞バリアントコーラー9には、いくつかの信頼できるオープンソースアルゴリズムの長所と、新しいDRAGENに特定のイノベーションが組み合わされています。全体的なアーキテクチャー(図7)はGATK 4.0のアーキテクチャーに基づいており、コアベイズジェノタイピングモデル(図5Aの「変異コール」コンポーネント)は、腫瘍/正常ワークフローの場合に腫瘍と正常サンプルの共同解析を行うStrelka2のアーキテクチャーに基づいています。DRAGENチームは、サンプル特異的なIndelエラー率とヌクレオチドエラーバイアスの推定(どちらもリード尤度計算を行うバックグラウンドのMarkovモデルのパラメーターに情報を与える)、ストランドバイアス、配向バイアス、ミスマッピングなどの一般的なエラーモードの確率モデルなど、いくつかの改善を追加しました。これらのイノベーションにより、上記の先行ツール(図8)の両方を上回る精度が得られました。
上述のように、DRAGENはペアの正常サンプルの存在下または非存在下での腫瘍サンプルの処理をサポートします。ペアの正常サンプルにより、DRAGENは生殖細胞系列バリアントをより精確に排除することができます。非存在下では、DRAGENは、1)公開されているデータセット(dbSNPまたはgnomADなど)の一般的なSNP、および2)その他の生殖細胞系列バリアント(プロキシフィルターとしても知られる)の近接性を活用して、生殖細胞系列バリアントを排除することができます。それにもかかわらず、このチャレンジでは、真の陽性体細胞の大部分が混合前に1つ以上の細胞株に生殖細胞系列バリアントとして存在していたため、参加者は生殖細胞系列バリアントを排除しないように指示を受けていました。
また、ペアの正常サンプルの存在は、DRAGENがPCRアーティファクト、ポリメラーゼのスリップ、G-クアドプレックスなどの二次構造の形成などから生じる系統的シーケンスノイズを特徴づけできるようにすることで、精度も向上させました。DRAGENパイプラインでは、正常サンプルが存在するかどうかにかかわらず(ただし、その非存在下では特に重要となる)、ユーザーが系統ノイズファイル(ノーマルパネルまたはPONとしても知られる)を生成して、正常サンプルパネルから位置特異的エラー率を推定することで、ラボおよびプロセス特異的ノイズ特性をキャプチャーすることもできます。これらのエラー率は、腫瘍サンプル内の特定の位置のシグナルがノイズとして統計的に説明できる場合、これらの位置のコールのフィルターに使用されます。*
*現在、ほとんどのパイプライン(DRAGENを含む)では、PONが使用されるときにSNVとIndel(異なるサイズのもの)を区別していません。しかし、このチャレンジでは、SNVエラーとIndelエラーを比較するために、新たな系統ノイズに対するアプローチが適用されました。つまり、精確なアレルのAFがPONからのサンプルに認められた場合、そのバリアントはフィルターされ、ペアではない正常サンプルのAFが<30%. If a variant has AF>30%であった場合、バリアントは維持されています(生殖細胞系列バリアントの可能性が高い)。概して、PONを使用すると、腫瘍パネルのシーケンスデータでFPを最大15%削減できることが社内ベンチマーキングデータから明らかになりました。
結論
このチャレンジは、イルミナのDRAGENチームがDRAGEN体細胞Indelコールパイプラインの能力を実証する機会となりました。DRAGENは精度で首位、適応性チャレンジではF1スコアで首位を獲得しました(パネルX)。DRAGENはまた、3種のパネルすべてを平均化した場合でも、最高のF1スコアを獲得し、3種のパネルで一貫して高い精度を示しました。これは、体細胞IndelコールにおけるDRAGENの高い性能が、単一のパラメーターセットを使用して複数の異なるパネルの一般化が可能であることを際立たせています。
学術用途向けの詳細情報またはDRAGEN試用版ライセンスについては、dragen-info@illumina.comまでお問い合わせください。
謝辞
PrecisionFDAと参加者の皆様の、このチャレンジを組織し、参加するための貴重な努力に感謝申し上げます。これまでのPrecisionFDAのチャレンジと同様に、今回もバイオインフォマティクスパイプラインの評価と最適化のために他に類を見ない環境が提供されました。また、過去および今回のチャレンジでDRAGENの開発に貢献したDRAGENチームのメンバー全員に感謝いたします。
注釈
1. GDC Data Portal with TCGA statistics
2. PrecisionFDA NCTR Indel Calling from Oncopanel Sequencing Data Challenge
4. DRAGEN Wins at PrecisionFDA Truth Challenge V2
5. Gong, B. et al. Cross-oncopanel study reveals high sensitivity and accuracy with overall analytical performance depending on genomic regions. Genome Biol 22, 109 (2021). https://doi.org/10.1186/s13059-021-02315-0
6. Sequencing benchmarked. Nat Biotechnol 39, 1027 (2021). https://doi.org/10.1038/s41587-021-01067-3
7. Jones, W. et al. A verified genomic reference sample for assessing performance of cancer panels detecting small variants of low allele frequency. Genome Biol 22, 111 (2021). https://doi.org/10.1186/s13059-021-02316-z
8. Ameur A: Goodbye reference, hello genome graphs. Nature biotechnology 2019, 37:866-868.
9. DRAGEN Somatic Small Variant Caller, Biorxiv, coming soon.