はじめに
近年、ヒトゲノム研究は、ゲノムの暗い領域を特徴付ける上で大きな課題に直面しています。これらの領域は、マッピング能力が低いため、組み立てやアライメントが困難で、マッピング可能なリードがほとんどないか、まったくなく、マッピング品質が低いアライメントされたリードになります。通常、これらの暗い領域は、高度に多型性または重複したゲノム領域に見られるため、依然として困難ですが、ヒトゲノムの理解を深める上で極めて重要な貴重な洞察を保持しています1,2
ヒトゲノム研究におけるもう1つの大きな課題はリファレンスバイアスです。これは、単一のハプロイドヒトリファレンスゲノムを使用して集団全体のヒトシーケンスの多様性を表現するという限界から生じています。3リファレンスバイアスはゲノムの特定の領域におけるリードマッピングの困難さを悪化させる可能性があるため、これらの2つの課題は相互に関連しています。GRCh38リファレンスアセンブリ4は、特に高度に多型な領域で、ヒトの遺伝的バリエーションをより精確にキャプチャーするために、FASTAリファレンスに代替(alt)コンティグをネイティブに含めることで、リファレンスバイアスの問題に少なくとも部分的に対処しようとしました。これらのネイティブな代替コンティグは、一次コンティグ内の対応する領域への代替経路を表しています。しかし、これらの追加コンティグの取り扱いには、コンティグが解決しようとしているエラーよりも多くのエラーが発生する可能性があるため、特別な注意が必要です。 これについては、以前のイルミナのGenomics Research Hubの記事5で説明しています。 この記事では、DRAGENが、マッピングの精度を向上させるためにGRCh38リファレンスで、これらのネイティブな代替コンティグと一次コンティグの関係を効果的に利用していることを示しています。当初は、Alt-awareアライメントのリフトオーバー手順を使用していましたが、これは後にAlt-maskingアプローチによってバージョン3.9で置き換えられ、マッピングの精度をさらに向上させました。
しかし、これらの限られた進歩では、ヒトの変異の大部分を完全にはキャプチャーしていません。これに対処するため、コミュニティーは現在、リファレンスとして役立つ高品質アセンブリの集合を作成しています 。この取り組みは、いくつかのパンゲノムコンソーシアムによって主導されています。Human Pangenome Reference Consortium(HPRC)、6 中国パンゲノムコンソーシアム(CPC)、7アラブのパンゲノムリファレンス(APR)、8 グローバルなリファレンス集合と祖先固有のリファレンスコレクションの両方の作成を目指しています。 HPRCや他のコンソーシアムの取り組みと並行して、DRAGEN はバージョン3.7で初めてマルチゲノム マッパーとパンゲノム リファレンスの概念を導入し、困難なゲノム領域におけるイルミナリードのマッピング精度を大幅に向上させました。9
注: これまでは、マッピングを実行する方法や、「マルチゲノム(グラフ)リファレンス」として使用されるサンプルの収集の両方の組み合わせについて言及してきました。今後、マッピングを行うための手法(マルチゲノムマッパー)と、マッピングのリファレンスとして使用するサンプルの集合(パンゲノムリファレンス)を分離し、各バージョンのDRAGENで導入された更新内容をより正確に説明できるようにします。
図1に示すように、DRAGENは時間の経過とともに、数世代のマルチゲノムマッパーとパンゲノムリファレンスをリリースし、反復するたびにマッピング困難領域におけるリードマッピングの精度がさらに向上しました。 図2は、マッピングに使用されるリファレンスの進化を示しています。
DRAGEN v3.7でリリースされた第1世代のマルチゲノムマッパーは、16の欧州サンプルで構成されるパンゲノムリファレンスから抽出された一連の集団ハプロタイプでネイティブの代替コンティグを拡張しました。第1世代マルチゲノムマッパーの導入により、線形リファレンス(ネイティブな代替コンティグを含む)の使用と比較して、SNPエラーが47%、Indelエラーが24%減少しました。
DRAGEN v4.2では、ヨーロッパ系の16サンプルから世界各地の 祖先を持つ32サンプルにパンゲノムリファレンスを拡大しました。これにより、祖先バイアスが低減し、バリアントコールの精度がさらに向上しました。
DRAGEN v4.3リリースでは、世界中の26の異なる祖先から32~128の集団サンプルに、当社のパンゲノムリファレンスを拡張できる第2世代マルチゲノムマッパーを導入しました(図3)。 このパンゲノム リファレンスは、DRAGENでこれまでに発売された最も多様な集団サンプルセットです。
本稿では、パンゲノムリファレンスを使用したマルチゲノムマッピング手法の進化について考察し、第2世代のマルチゲノムマッパーと最新の128サンプル パンゲノムリファレンスを用いて、ゲノムの全ベンチマーク領域とマッピング困難領域の両方で、有意な精度の向上を示しています。
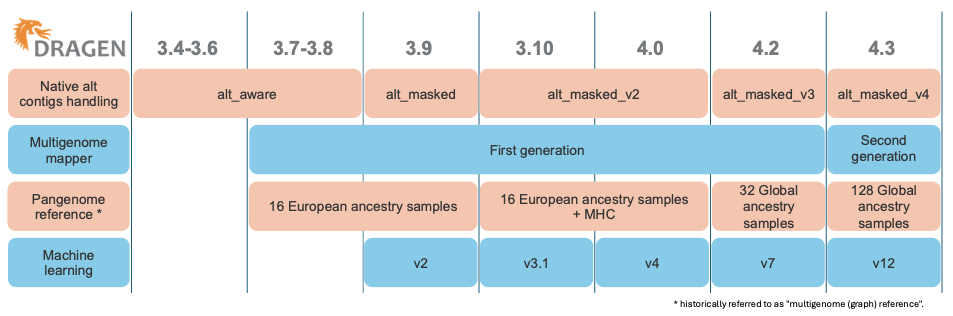
図1:DRAGENコンポーネントの進化には、ネイティブな代替コンティグの処理、マルチゲノムマッパー、パンゲノムリファレンス、機械学習が含まれます。
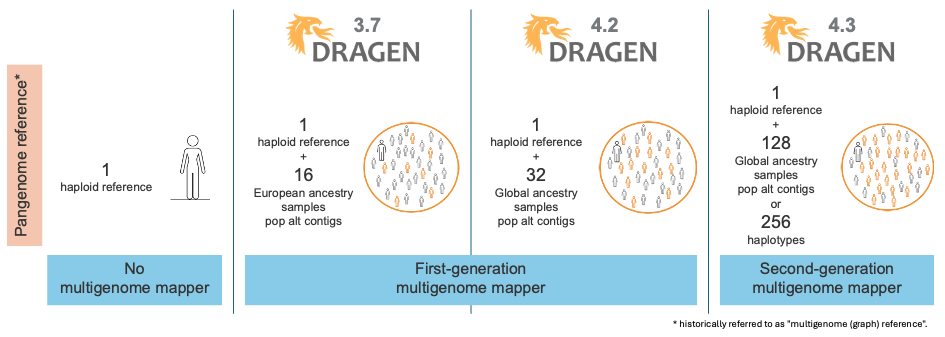
図2:DRAGENマルチゲノムマッパーの進化:集団認識機能およびパンゲノムリファレンス。
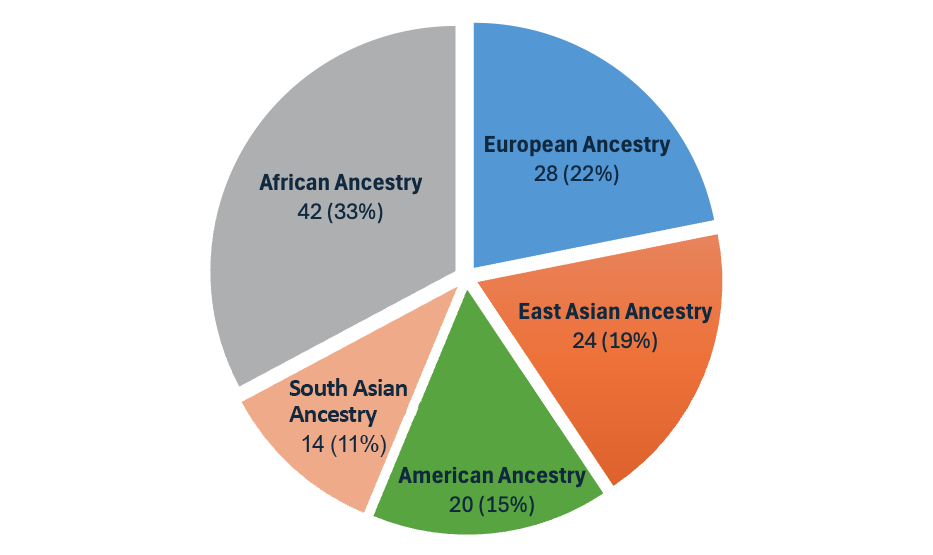
図3:DRAGEN v4.3のパンゲノムリファレンスは、世界中の26の祖先からの128の集団サンプルで構成されています。
DRAGEN v4.3は、史上最も精確なDRAGEN解析を実現します
図4は、SNPとIndelの組み合わせについて、v4.2.1 全ベンチマーク領域10のHG002 NISTサンプルすべてにおけるDRAGENの連続するバージョンの精度を報告しています。 DRAGENの精度は時間の経過とともに大幅に向上し、過去4年間で83%のエラー減少を達成しました。 第1世代マルチゲノムマッパーと16サンプルパンゲノムリファレンス をバージョン3.7.5で導入したことで、最初の急激なエラー減少につながりました。DRAGEN v4.3に第2世代 マルチゲノムマッパーと128のサンプルパンゲノムリファレンスが導入されたことで、DRAGENは精度の新たな基準を設定し続け、DRAGEN v4.2と比較してさらに40%の急激なエラー削減を達成しました。
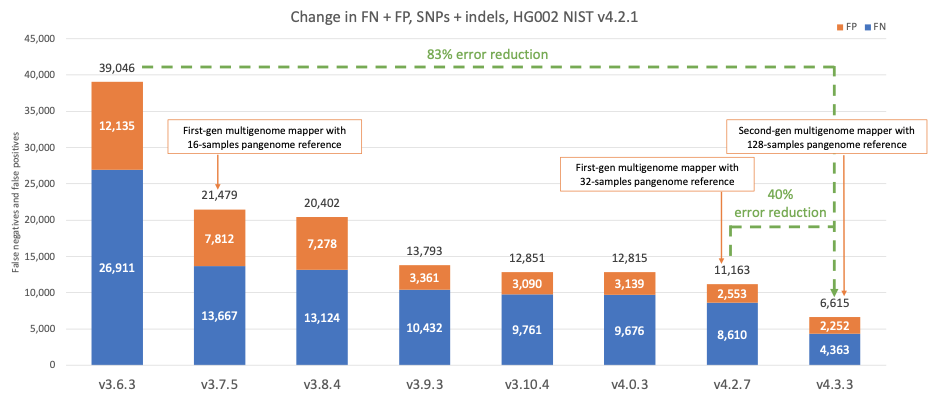
図4:HG002 NIST v4.2.1 SNPおよびIndel偽陽性と偽陰性エラーは、連続するDRAGENバージョンでカウントされます。
NIST v4.2.1の真理値セットを使用したGIABサンプルのベンチマーキング
図5では、DRAGEN v4.3の精度をDRAGEN v4.2と比較し、アライメントにはGiraffe11 1.54.0、すべてのベンチマーク領域でGenome in a Bottle(GIAB)13 v4.2.1を使用したHG001–HG007サンプルの小規模バリアントコールにはDeepVariant12 1.6.0で構成されるサードパーティパイプラインを比較しています。14Giraffe-DeepVariantは、SNPとIndelの組み合わせで平均エラー減少率61.61%、SNPで平均エラー減少率63.8%、Indelで53.53%を示しています。
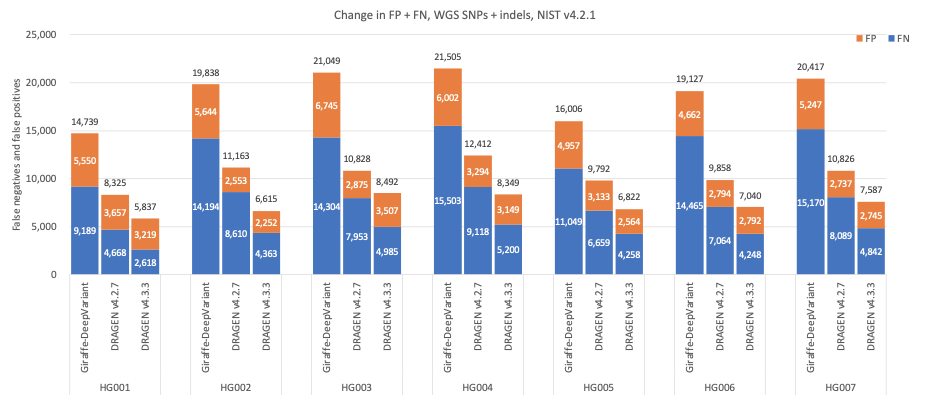
図5:DRAGEN v4.2、DRAGEN v4.3、およびGiraffe-DeepVariantでは、NIST v4.2.1真理値セットを使用して、7つのGIABサンプル(HG001~7)のSNPとIndelの累積エラーがカウントされます。
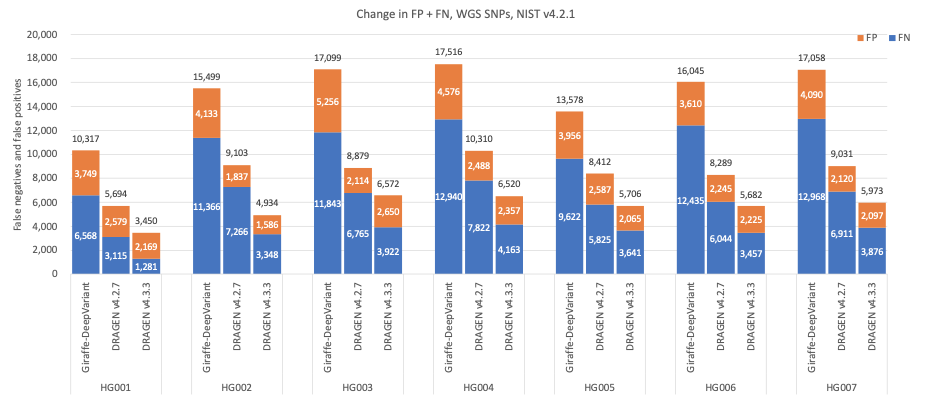
図6:DRAGEN v4.2、DRAGEN v4.3、およびGiraffe-DeepVariantについて、NIST v4.2.1の真理値セットを使用した7つのGIABサンプル(HG001~7)のSNP累積エラー数。
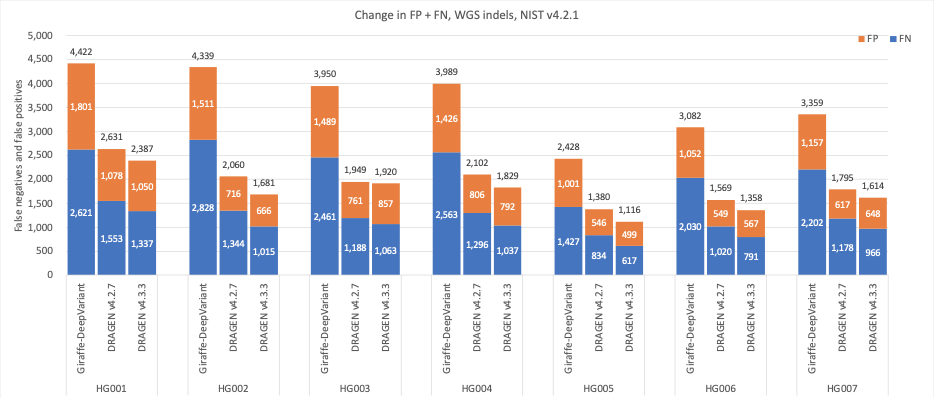
図7:DRAGEN v4.2、DRAGEN v4.3、およびGiraffe-DeepVariantについて、NIST v4.2.1の真理値セットを使用した7つのGIABサンプル(HG001~7)のIndel累積エラー数。
ゲノムのマッピング困難領域におけるGIABサンプルのベンチマーキング
図8は、NIST15,16で定義されているように、マッピング困難領域における精度の結果を示しています。DRAGEN 4.3ではSNPおよびIndelの精度に目覚ましい増加が見られ、Giraffe-DeepVariantと比較して平均エラー減少率は65.51%、DRAGEN 4.2と比較して37.77%でした。
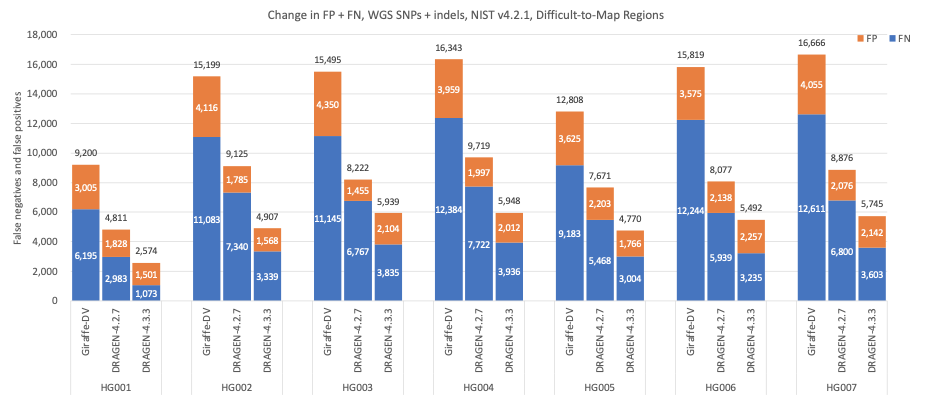
図8:DRAGEN v4.2、DRAGEN v4.3、およびGiraffe-DeepVariantのマッピング困難領域におけるBEDファイルと交差するNIST v4.2.1真理値セットを使用した、7つのGIABサンプル(HG001~7)のSNPおよびIndelの累積エラー数。
CMRG真理値セットを用いたHG002サンプルのベンチマーキング
図9は、SNP とIndelを組み合わせた、Challenging Medically Relevant Genes (CMRG) の真理値セット17における連続したDRAGENバージョンの精度を示しています。合計エラー数は時間の経過に伴い継続的に減少しており、これはNIST v4.2.1 全ベンチマーク領域におけるエラー数の変化と一致しています。DRAGEN v4.3による128のサンプルパンゲノムリファレンスを持つ最近の第2世代マルチゲノムマッパーは、偽陽性と偽陰性の合計をさらに15%減少させ、複数の遺伝子にわたって85の偽陰性コールを精確に検出しました。DRAGEN v4.2とDRAGEN v4.3はどちらも、Giraffe-DeepVariantパイプラインを上回っています。Giraffe-DeepVariantと比較すると、DRAGEN v4.3は医学的に関連する遺伝子の200を超える追加バリアントを精確に検出します。
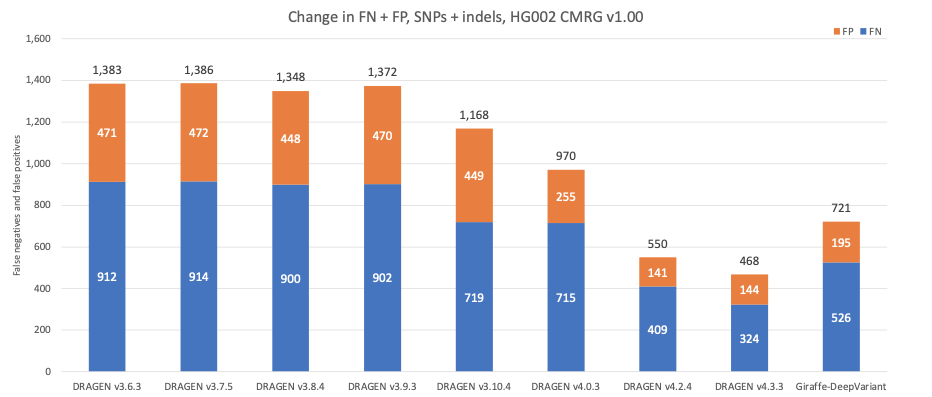
図9:HG002 SNPとIndelの組み合わせ精度により、連続するDRAGENバージョンとGiraffe-DeepVariantで設定されたCMRG真理値セットが得られます。
DRAGEN第1世代マルチゲノムマッパー法
DRAGENマルチゲノムマッパーの第1世代では、パンゲノム標準サンプルのフェーズ済みSNPおよびIndelコールから、ゲノムのマッピング困難領域における集団ハプロタイプを導き出しました。この際、信頼度の低いバリアントや集団アレル頻度の低いバリアントは除外されています。特に、16個のヨーロッパの祖先サンプルから32個の集団ハプロタイプを導き出し、32個の世界の祖先サンプルから64個の集団ハプロタイプを導き出しました。
DRAGEN第2世代マルチゲノムマッパー手法
DRAGEN v4.3に導入された第2世代のマルチゲノムマッパーは、第1世代の表現に基づいて構築され、その主な限界に対処します。利用可能な集団アセンブリの数が増えるにつれ、第2世代マルチゲノムマッパーは、垂直方向(集団パンゲノムのサンプル数)と水平方向(全ゲノムカバレッジ)の両方のスケーラビリティを向上させるように設計されています。さらに、すべての集団バリアントがフェーズされることを保証する、長距離フェージングを可能にする新しい構造に依存しています。
このデータ構造では、独自の圧縮法を使用して、複数のハプロタイプ間で共有される冗長バリアント情報の保存を回避し、ローカルで異なるハプロタイプのセットのみを記録しながら、すべての長距離情報を保持します。このアプローチにより、より多くのハプロタイプを追加する際に効率的なスケーリングが可能になり、スコアリング速度が向上します。ハプロタイプパネルのサイズは、より多くの祖先を含むDRAGEN v4.2の64からDRAGEN v4.3の256へと大幅に増加しました。
特に、マルチ塩基コードとしてフェージングされていないSNPを含む第1世代と比較して、この新しいバージョンはすべてのバリアントをフェーズし、ハプロタイプ全体でペアエンドメイトのより精確なジョイントスコアリングを可能にします。
代替コンティグ用の以前のリフトオーバーシステムはまだ使用されていますが、現在では複雑なバリアントに焦点を合わせています。集団SNPは、種子マッピングプロセスの感度を向上させるため、一次リファレンスでは依然としてマルチ塩基コードとしてエンコードされますが、アライメントスコアは、新しいデータ構造からの段階的なバリアント情報を使用して計算されます。
コマンドラインオプション
バージョン4.3.3より前のDRAGENランに使用されるコマンドラインオプションは、以前のイルミナのGenomics Research Hubの記事で報告されています。19 DRAGEN v4.3.3ランでは、表1にも説明されている以下のコマンドラインオプションを使用しました。
dragen \
--fastq-file1 <path-to-R1-fastq> \
--fastq-file2 <path-to-R2-fastq> \
--RGSM HG002 \
–RGID HG002 \
--ref-dir <path-to-reference-directory> \
--output-file-prefix HG002 \
--events-log-file Dragen_events.csv \
--output-directory <path-to-output-directory> \
--generate-sa-tags true \
--enable-vcf-compression true \
--enable-variant-caller true \
--enable-map-align true \
--enable-map-align-output true \
--enable-sort true \
--enable-duplicate-marking true \
--enable-bam-indexing true

表1:テストで使用したDRAGENコマンドラインオプション。
DRAGEN v4.3 hg38-alt_masked.graph.cnv.hla.rna_v4 ハッシュテーブルはDRAGEN製品ファイルページ20 から入手可能で、DRAGEN v4.3 MLモデルv12.0は初期設定であり、DRAGEN実行可能ファイルとともにパッケージ化されています。
NISTの基準値との一致率は、Nature Biotechnology21に記載されているRTGツールキットを使用して実行されます。比較例のコマンドは、次のとおりです。
java \
-Djava.awt.headless=true \
-Dtalkback=false \
-Dusage=false \
-Xmx40g \
-jar RTG-3.9.1.jar vcfeval \
-b <truth>.vcf.gz \
-c <query>.vcf.gz \
-t <tmp_dir> \
-o <output_dir> \
--output-mode annotate \
--vcf-score-field QUAL \
--bed-regions <truth>.bed \
-Z \
--sample <truth sample>,<query sample> \
--ref-overlap
すべてのDRAGENランで、推奨される<sample>.hard-filtered.vcf.gz VCF出力ファイルを使用します。これにより、最良のf1スコア測定値が得られます。
マッピング困難領域を評価する際、RTGツールキットに層別化ベッドを提供するために、--evaluation-regions <stratification>.bedフラグを追加しました。
Giraffeベースのパイプラインについては、AWSでリリースされたhprc-v1.1-mc-grch38.d9リファレンスを使用して、DeepVariant-VGケーススタディ22で指定されたレシピに従っていました。23 Giraffe v1.52.0を使用してリードをアライメントしました。コマンドラインとパラメーターは以下のとおりです。
vg giraffe \
--progress \
--read-group "ID:1 LB:lib1 SM:HG002 PL:illumina PU:unit1" \
--sample "HG002" \
--prune-low-cplx \
--max-fragment-length 3000 \
--output-format bam \
-f <path-to-R1-fastq> \
-f <path-to-R2-fastq> \
-x hprc-v1.1-mc-grch38.d9.xg \
-Z hprc-v1.1-mc-grch38.d9.gbz \
-d hprc-v1.1-mc-grch38.d9.dist \
-m hprc-v1.1-mc-grch38.d9.min \
--ref-paths hprc-v1.1-mc-grch38.d9.ref_paths \
-t 32 > HG002.giraffe.grch38.d9.bam
出力BAMをsambamba v0.8.1で並べ替え、samtools v1.15.1でインデックスします。
sambamba sort \
-t 32 \
-o HG002.giraffe.grch38.d9.sort.bam \
HG002.giraffe.grch38.d9.bam
samtools index \
-@ 32 \
HG002.giraffe.grch38.d9.sort.bam
次のsingularityコマンドで、DeepVariant v1.6.0を使用したバリアントコール:
singularity run \
--bind "${INPUT_DIR}:/mnt/input,${REF_DIR}:/mnt/reference,${OUTPUT_DIR}:/mnt/output,${BIND_TMPDIR}:/tmp" \
deepvariant_1.6.0.sif \
/opt/deepvariant/bin/run_deepvariant \
--ref="/mnt/reference/hprc-v1.1-mc-grch38.d9.fa" \
--reads="/mnt/input/HG002.giraffe.grch38.d9.sort.bam" \
--model_type="WGS" \
--sample_name="HG002" \
--output_vcf="/mnt/output/HG002.vcf.gz" \
--output_gvcf="/mnt/output/HG002.g.vcf.gz" \
--make_examples_extra_args="min_mapping_quality=1,keep_legacy_allele_counter_behavior=true,normalize_reads=true" \
--haploid_contigs="chrX,chrY" \
--par_regions_bed="/mnt/reference/hprc-v1.1-mc-grch38.d9.par_regions.bed" \
--num_shards="40"
まとめ
DRAGENは、さまざまなリリースを通じて、コンポーネントを綿密に設計することで、精度について新しい基準を継続的に設定 してきました。この記事では、パンゲノムリファレンスに基づく新世代のDRAGENマルチゲノムマッピング(第2世代)に焦点を当てていますが、機械学習やバリアントコーリングなどのその他のコンポーネントもDRAGEN v4.3で強化されており、その高性能に貢献しています。
DRAGEN v4.3の進歩の詳細については、イルミナソフトウェアリソースブログをご覧ください。
全体として、DRAGEN v4.3はゲノムの暗い領域でも生殖系列の小規模バリアントを呼び出すのに驚異的な精度を示し、NIST v4.2.1 全ベンチマーク領域およびマッピング困難領域、およびCMRG領域でのテストにおいて、一貫して以前のバージョンとサードパーティのパイプラインを上回っています。精確性、スピード、スケーラビリティ、遍在性、包括性で知られるDRAGENは、ゲノム解析に不可欠なツールであり、研究者やサイエンティストに包括的で効率的なゲノムデータ解析のための強力なリソースを提供します。
学術用途向けの詳細情報またはDRAGEN試用版ライセンスについては、dragen-info@illumina.comまでお問い合わせください。
注釈
- Ebbert MTW, Jensen TD, Jansen-West K, et al. Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight. Genome Biol . 2019;20(1):97. doi:10.1186/s13059-019-1707-2
- Ryan NM, Corvin A. Investigating the dark-side of the genome: a barrier to human disease variant discovery? Biol Res . 2023;56(1):42. doi:10.1186/s40659-023-00455-0
- Miga KH, Wang T. The Need for a Human Pangenome Reference Sequence. Annu Rev Genomics Hum Genet . 2021;22:81-102. doi:10.1146/annurev-genom-120120-081921
- GRCh38 Full Analysis Set Plus Decoys HLA. ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/GRCh38_reference_genome/GRCh38_full_analysis_set_plus_decoy_hla.fa
- Catreux S, Farrell F, Mehio R, et al. Demystifying the versions of GRCh38/hg38 reference genomes, how they are used in DRAGEN and their impact on accuracy. Illumina website. illumina.com/science/genomics-research/articles/dragen-demystifying-reference-genomes.html. Published December 9, 2021. Accessed July 18, 2024.
- Liao WW, Asri M, Ebler J, et al. A draft human pangenome reference. Nature. 2023;617(7960)312-324. doi:10.1038/s41586-023-05896-x
- Gao Y, Yang X, Chen H, et al. A pangenome reference of 36 Chinese populations. Nature. 2023;619(7968):112-121. doi:10.1038/s41586-023-06173-7
- Uddin M, Nassir N, Almarri M, et al. A draft Arab pangenome reference. Preprint. Research Square. October 2023. doi:10.21203/rs.3.rs-3490341/v1
- Catreux S, Jain V, Murray L, et al. DRAGEN sets new standard for data accuracy in PrecisionFDA benchmark data. Optimizing variant calling performance with Illumina machine learning and DRAGEN graph. Illumina website. illumina.com/science/genomics-research/articles/dragen-shines-again-precisionfda-truth-challenge-v2.html. Published January 12, 2022. Accessed July 18, 2024.
- GIAB HG002 v4.2.1 truth. ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/AshkenazimTrio/HG002_NA24385_son/NISTv4.2.1/.
- Sirén J, Monlong J, Chang X, et al. Pangenomics enables genotyping of known structural variants in 5202 diverse genomes. Science. 2021;374(6574). doi:10.1126/science.abg8871
- Poplin R, Chang PC, Alexander D, et al. A universal SNP and small-Indel variant caller using deep neural networks. Nat Biotechnol. 2018;36(10):983-987. doi:10.1038/nbt.4235
- Wagner J, Olson ND, Harris L, et al. Benchmarking challenging small variants with linked and long reads. Cell Genom . 2022;2(5):100128. doi:10.1016/j.xgen.2022.100128
- GIAB samples release. ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/.
- Difficult-to-Map Regions BED file. ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/genome-stratifications/v3.3/GRCh38@all/Union/GRCh38_alllowmapandsegdupregions.bed.gz.
- Olson ND, Wagner J, McDaniel J, et al. PrecisionFDA Truth Challenge V2: Calling variants from short and long reads in difficult-to-map regions. Cell Genom . 2022;2(5):100129. doi:10.1016/j.xgen.2022.100129
- Wagner J, Olson ND, Harris L, et al. Curated variation benchmarks for challenging medically relevant autosomal genes. Nat Biotechnol . 2022;40(5):672-680. doi:10.1038/s41587-021-01158-1
- Behera S, Catreux S, Rossi M, et al. Comprehensive and accurate genome analysis at scale using DRAGEN accelerated algorithms. Nat Biotechnol. 2024. Published 2024 Oct 25. doi:10.1038/s41587-024-02382-1
- Rossi M, Catreux S, Roddey C, et al. Unlocking the full potential of Illumina genomes: The journey to enhanced variant calling quality with DRAGEN informatics and high-quality sequencing. Illumina website. illumina.com/science/genomics-research/articles/CMRG_hg38.html. Published June 29, 2023. Accessed July 18, 2024.
- DRAGEN Bio-IT Platform Product Files. emea.support.illumina.com/sequencing/sequencing_software/dragen-bio-it-platform/product_files.html.
- Krusche P, Trigg L, Boutros PC, et al. Best practices for benchmarking germline small-variant calls in human genomes. Nat Biotechnol. 2019;37(5):555-560. doi:10.1038/s41587-019-0054-x
- Using graph genomes: VG Giraffe + DeepVariant case study. github.com/google/deepvariant/blob/0bba6a71b8b0c2046a3b01c0bda1a5a0d2b80fca/docs/deepvariant-vg-case-study.md.
- hprc-v1.1-mc-grch38. s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=pangenomes/freeze/freeze1/minigraph-cactus/hprc-v1.1-mc-grch38/.