ファクトシート
- ゲノムバリアントのアノテーションは複雑なプロセスです
- この分野で使用されている一般的なツールには、精度やスケーリングの制限があります
- Emedgeneに新たに追加され、DRAGEN二次解析で使用可能なIllumina Connected Annotationsは、この分野で卓越した性能を発揮します
- 『All of Us』や『UK Biobank』などのトップクラスの人口統計調査は、このツールに依存しています
背景:
すべての人に存在する数百万の遺伝的バリアントを確実に検出する能力は、全ゲノムシーケンスの使用において飛躍的に増加しました。これらすべてのバリアントを理解するには、遺伝子モデル、集団内頻度、インパクト予測、リファレンスデータベースなどの有用な情報でアノテーションする必要があります。遺伝性遺伝子スクリーニングから腫瘍学や集団遺伝子研究まで、処理されたデータに正確かつ迅速にアノテーションを行えることが、ゲノミクスプロジェクトの成功の鍵となります。
これまでに構想された最も意欲的な集団ゲノミクスプログラムである「All of Us Research Program」と「UK Biobank project」は、どちらも途方もない規模のアノテーションの課題に直面しました。何十万人もの個人と10億を超えるユニークなゲノムバリアントに注釈を付けるため、彼らは、効率性と正確性に関する非常に高い制度スタンダードを満たすソフトウェアを必要としていました。複数のオプションを評価した後、両グループは、イルミナのアノテーターを自分たちのニーズを満たす独自の機能を備えているとして、選択ツールとして採用しました。1,2 最近のリリースでは、UK Biobankは、約90分で500,000の全ゲノムマルチサンプルバリアントコールファイルのデータセット全体にアノテーションを成功させました。3
イルミナのシーケンサーを使用するサイエンティストに力を与えるため、当社のバイオインフォマティクスチームは長年にわたり、アノテーションのニーズに対処するソフトウェアを開発してきました。また、当社の最新バージョンであるIllumina Connected Annotationsをリリースできることを誇りに思います。Illumina Connected Annotationsは、以前のNIRVANAというツールを基盤に構築されており、Illumina Connected Softwareスイートの一部です。この最先端のゲノムアノテーションエンジンは、既存のイルミナソフトウェアソリューションのスピードと精度を組み合わせ、イルミナの主要な解析および解釈製品(DRAGEN4やEmedgeneなど)のコアコンポーネントとして直接統合することで、ブレークスルーな探索に必要な柔軟性とカスタマイズを可能にします。
Illumina Connected Annotationsが提供するものとは?
アノテーションソフトウェアは、DRAGEN DNAおよびRNA解析パイプラインによって生成されるような小規模バリアント(複数塩基変異を含む)、コピー数バリアント(CNV)、SV、STR、および/または融合遺伝子の単一またはマルチサンプルVCFで読み取ります。
Illumina Connected Annotationsは、Human Genome Variation Society(HGVS)スタンダードに従って、コーディングシーケンス(「c.」または「cNomen」)およびタンパク質シーケンス(「p.」または「pNomen」)で予測される影響だけでなく、5 Sequence Ontology スタンダードの命名法を使用した各バリアントに関連する結果を提供します。6転写および遺伝子モデルは、RefSeq(バージョンGCF_000001405.40-RS_2023_03, 2023-03-21)およびEnsembl(リリース110, 2023-04-27)から取得されます。
機能アノテーションの仕組みとは?
Illumina Connected Annotationsの一部としての機能アノテーション
- インターバルアレイを使用して、各代替対立遺伝子と交差する転写産物を同定し、
- 重複するエクソンおよび/またはイントロンをマークし、
- 転写産物とゲノムリファレンスシーケンス間の不一致を調整し、
- cNomenとpNomenを提供します。
カノニカル転写産物は、MANE7からの情報、または入手できない場合は既存のヒューリスティック手法を用いて同定されます。8,9 このフェーズでは、シーケンス腫瘍学の標準命名法を使用して、各バリアントに関連する結果も提供します。Illumina Connected Annotationsは、HGVSスタンダードに従って、必要に応じてコーディングおよびタンパク質シーケンスに対して右揃えを実行します。5該当する場合、関連するがんホットスポットのアノテーションも追加します。次に、得られた遺伝子の方向性と近接性に基づいて一方向遺伝子融合の可能性を同定するためにSVを評価します。既知の融合遺伝子は、COSMIC10やFusionCatcherなどのリソースの対合遺伝子記号を使用してアノテーションされます。11最終段階では、VCFに少なくとも1つのバリアントを持つ各固有遺伝子に遺伝子固有のアノテーションを追加し、OMIM、ClinGen、およびその他の遺伝子情報ソースからデータを取得します。
アノテーションソース
また、Illumina Connected Annotationsは、gnomADやCOSMICなどの最も一般的に使用されている強力な公開データベースだけでなく、SpliceAI12やPrimateAI-3Dなどイルミナで開発された予測スコアを統合しています。13ブレークスルーな探索のためのゲノムAIのパワーとイルミナがどのように道を切り開くかについては、この記事のGenetic Engineering & Biotechnology Newsでご覧ください。
サポートされているデータソースは表1にリストされており、2つの階層に分かれています。プレミアムおよびベーシック。Basicティアには無料でアクセスできます。Premiumティアにはライセンスが必要です。アクセスについては、annotation_support@illumina.comまでお問い合わせください。
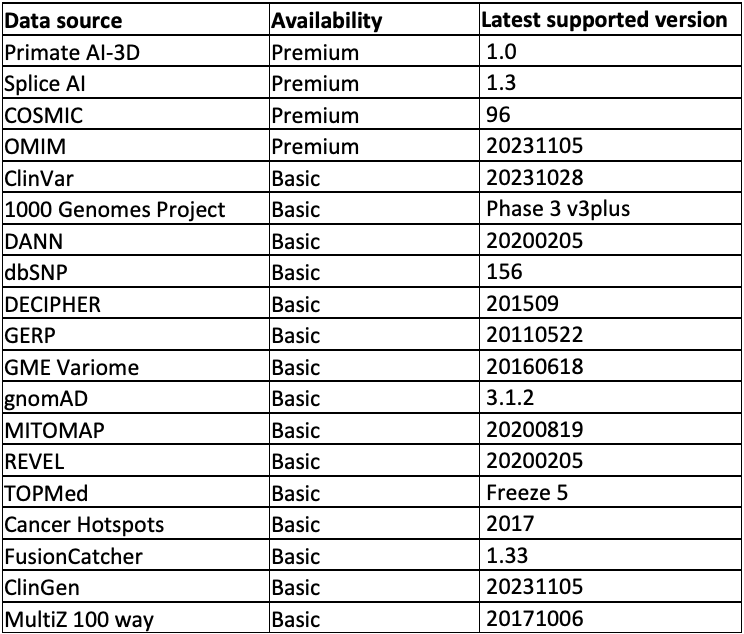
表1:Illumina Connected Annotationsツールですぐに利用できるアノテーションソース:上記のデータソースに加えて、自分のデータを含むほとんどのアノテーションリソースをカスタムアノテーションリソースにフォーマットできることにご注意ください。この表は、ここで最新の状態に保たれています。
精度とスピード
精度を測定するため、全ゲノムシーケンス(WGS)および全エクソームシーケンス(WES)から呼び出された900万近いバリアントのセットについて、Illumina Connected Annotationsの出力とHGVSの結果を比較しました。また、サードパーティ製ツールVEP、14 SnpEff、15およびANNOVARと精度を比較しました。16
この実験では、各アノテーションツールで生成されたHGVS表記と、ゴールドスタンダードとして認識されているBioCommonsが提供するHGVS表記を直接比較しました。
図1は、WGS VCF(約650万のバリアント)のHGVSゲノム、コーディング、およびタンパク質表記のIllumina Connected Annotations、VEP、SnpEff、およびANNOVARの精度結果を示しています。
- HGVSゲノム精度:イルミナのConnected Annotationsは、すべてのバリアントに正しくアノテーションを付け、100%の精度を達成し、VEPは99.970%の精度を示しています
- HGVSコーディング精度:イルミナのConnected Annotationsの精度は99.997%で、その他のツールは99.962~99.991%です。
- HGVSタンパク質の精度:イルミナのConnected Annotationsは、99.998%という高い精度を示し、VEPの精度と一致し、99.988%を超える精度率を持つSnpEffとANNOVARをわずかに上回っています。
全体として、イルミナのConnected Annotationsは、HGVS表記のすべてのカテゴリーで高い精度を示します。
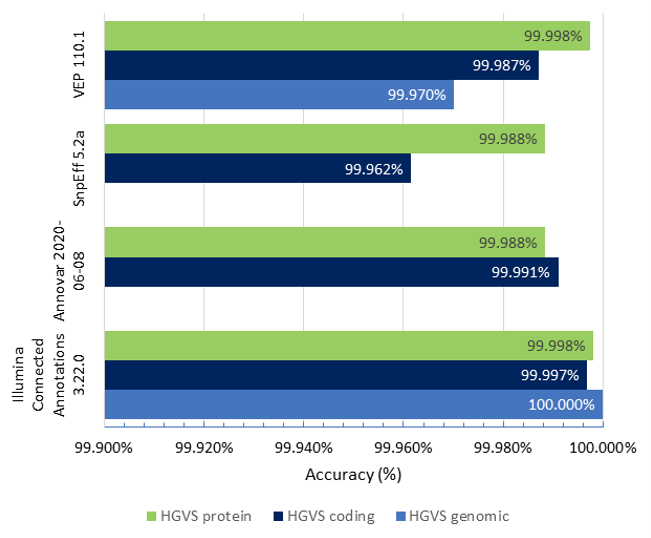
図1:BioCommonsがリファレンスとして提供するHGVS表記を用いたWGS VCFのHGVSゲノム、コーディング、タンパク質表記に対するIllumina Connected Annotations、VEP、SnpEff、ANNOVARの正確性。
Illumina Connected Annotationsは、生殖系列遺伝学と体細胞腫瘍学の両方のアプリケーションにおいて、集団規模で全ゲノム解析に高度に最適化されています。典型的なヒトゲノムには400万以上のバリアントがあり、腫瘍ゲノムにはそれよりもはるかに多くのものがあるため、アノテーション速度が大きなボトルネックになる可能性があります。
Illumina Connected Annotationsがどのように機能するかを示すために、約650万のバリアントを含む全ゲノム生殖系列VCFのアノテーションを実行しました。また、比較としてVEP、SnpEff、ANNOVARを使用して同じVCFファイルにアノテーションを実行しました。以下の図2は、各ソフトウェアが約650万のバリアントにアノテーションを行うランタイムを示しています。
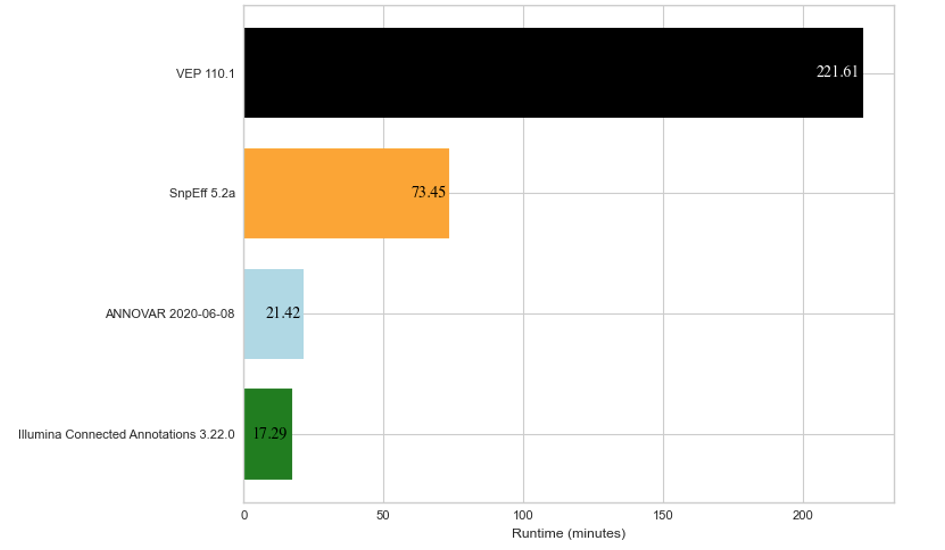
図2:4つの異なるアノテーションアルゴリズムの3つのレプリケートにわたる約650万のDNAバリアントのアノテーションの平均ランタイム結果。
上記で比較したアノテーションは、それぞれ同じクラウドベースのハードウェア(AWS EC2 c5.4xlarge with 16vCPU and 32 GB of RAM)で実行されました。SnpEffは、RefSeqデータベースのみを使用して注釈を付けますが、VEP、ANNOVAR、Illumina Connected Annotationsは、RefSeqデータベースとEnsemblデータベースの両方を使用することにご注意ください。
集団ゲノミクス研究へのスケーリング
Illumina Connected Annotationsは、DRAGEN Iterative gVCF Genotyperで生成された単一サンプルVCFだけでなく、マルチサンプルVCFにも注釈を付けることができ、大規模な注釈付けが可能です。大量のサンプルレベルの情報をアノテーション出力JSONファイルにコピーすることを避けるために、ユーザーはオプションで、マルチサンプルVCFから抽出された必須情報のみを含むサイトレベルのVCF、すなわちCHROM、POS、REF、およびALT(ALTには複数のアレルが含まれる場合があります)を作成することができます。このサイトレベルのVCFは、Illumina Connected Annotationsへの入力として使用でき、出力ファイルでは、すべてのコホートサンプルで呼び出されたバリアントが一度だけアノテーションされます。ユーザーは、注釈プロセスの並列化を最大化するために、サイトレベルのVCFをゲノム領域ごとにさらに分割することができます。この方法を用いて、The 1000 Genomes Project(3,202サンプル)で検出された1億2,800万のバリアントと、UK Biobank 500,000全ゲノムリリース(490,541サンプル)で検出された15億のバリアントに注釈を付けました。2,3また、Illumina Connected Annotationsは、All of Us Research Programにおける10億以上の遺伝的バリアントのジョイントコールセットの注釈付けにも使用されました。1
ラン方法
下の表2に示すように、Illumina Connected Annotationsはスタンドアロンの実行可能ファイルとして、またはDRAGEN二次解析内で、さまざまな使用例で実行できます。これには、Illumina Connected AnalyticsとBaseSpace Sequence Hubを使用したクラウドベースの実行、およびオンプレミスサーバーが含まれます。DRAGENユーザー向けのIllumina Connected Annotations内のプレミアムティアデータソースの使用は無料です。

表2:Illumina Connected Annotationsのデプロイメントモデル:*Premiumデータベースのライセンスを取得するには、annotation_support@illumina.comまでEメールでお問い合わせください。†スタンドアロンビルドはこちらからダウンロードしてください。‡DRAGENバージョン4.3で利用可能。
ゲノム革命にご参加ください
ゲノミクス分野では、Illumina Connected Annotationsとその機能について、基本的かつプレミアムなデータソースで探求してください。私たちと共に、可能性の限界を押し広げましょう。Illumina Connected Annotationsは単なるツールではありません。これは進歩のきっかけであり、ゲノム研究への取り組みにどのような変革をもたらすのか、当社も待ちきれない思いです。
詳細を知りたい場合、ツールを使用したい場合、または将来のバージョンに組み込むべき追加リソースの提案が必要な場合は、annotation_support@illumina.comまでご連絡ください。ツールのスタンドアロンバージョンはこちらからダウンロードでき、ドキュメントはこちらから入手できます。
学術用途向けの詳細情報またはDRAGEN試用版ライセンスについては、dragen-info@illumina.comまでお問い合わせください。
注釈
- The All of Us Research Program Genomics Investigators. Genomic data in the All of Us Research Program. Nature. February 19, 2024. doi:10.1038/s41586-023-06957-x
- Halldorsson BV, Eggertsson HP, Moore KHS, et al. The sequences of 150,119 genomes in the UK Biobank. Nature. July 2022;607(7920):732-740. doi:10.1038/s41586-022-04965-x
- Science. UK Biobank releases half a million whole-genome sequences for biomedical research. science.org/content/article/uk-biobank-releases-half-million-whole-genome-sequences-biomedical-research. November 29, 2023. Accessed February 2024.
- Behera S, Catreux S, Rossi M, et al. Comprehensive and accurate genome analysis at scale using DRAGEN accelerated algorithms. Nat Biotechnol. 2024. Published 2024 Oct 25. doi:10.1038/s41587-024-02382-1
- den Dunnen JT. Describing Sequence Variants Using HGVS Nomenclature. Methods Mol Biol. 2017;1492:243-251. doi:10.1007/978-1-4939-6442-0_17
- Eilbeck K, Lewis SE, Mungall CJ, et al. The Sequence Ontology: a tool for the unification of genome annotations. Genome Biol. 2005;6(5):R44. doi:10.1186/gb-2005-6-5-r44
- Chaisson MJP, Sanders AD, Zhao X, et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun. April 16, 2019;10(1):1784. doi:10.1038/s41467-018-08148-z
- Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. January 4, 2016;44(D1):D862-D868. doi:10.1093/nar/gkv1222
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. May 2015;17(5):405-424. doi:10.1038/gim.2015.30
- Bamford S, Dawson E, Forbes S, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. July 19, 2004;91(2):355-358. doi:10.1038/sj.bjc.6601894
- Beccuti M, Carrara M, Cordero F, et al. Chimera: a Bioconductor package for secondary analysis of fusion products. Bioinformatics. December 15, 2014;30(24):3556-3557. doi:10.1093/bioinformatics/btu662
- Jaganathan K, Panagiotopoulou SK, McRae JF, et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. January 24, 2019;176(3):535-548 e24. doi:10.1016/j.cell.2018.12.015
- Gao H, Hamp T, Ede J, et al. The landscape of tolerated genetic variation in humans and primates. Science. June 2, 2023;380(6648):eabn8153. doi:10.1126/science.abn8197
- McLaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol. June 6, 2016;17(1):122. doi:10.1186/s13059-016-0974-4
- Cingolani P. Variant Annotation and Functional Prediction: SnpEff. Methods Mol Biol. 2022;2493:289-314. doi:10.1007/978-1-0716-2293-3_19
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. September 2010;38(16):e164. doi:10.1093/nar/gkq603