元の説明
Guillaume Duchenne de Boulogne氏は、19世紀で最も影響力のある医学サイエンティストの1人として記憶されるべきであり、彼はキャリアを通して神経学に先駆的な貢献をしました。彼は、最初に筋生検を行い、臨床目的で写真を使用した人であり、神経伝導検査を行う装置を発明しました。
彼の治療を受けている患者の中には、特に壊滅的な筋肉の喪失に苦しむ一連の少年がいました。これは歩行困難から始まり、最終的には横隔膜の筋肉構造の不全により死亡しましたが、この筋肉構造の不全により、呼吸を妨げられていたのです。この疾患は、Duchenne氏が1861年にそのストーリーを含む本を発表するまで広く認識されていませんでした。この疾患には、彼の名前にちなんで名付けられています。デュシェンヌ型筋ジストロフィー。
DMD関連疾患のクリニカルな症状と自然経過
DMDは、知られている最大のヒト遺伝子の1つで、細胞帯Xp21.2-p21.1のX染色体にあります。これはジストロフィンをコードしており、ジストロフィンは、細胞骨格と細胞外マトリックス間の結合を維持し、それによって筋細胞膜の完全性を維持する上で重要な役割を果たす、ジストロフィン関連糖タンパク質複合体の一部です。1
DMDに関連する以下の3つの異なる症状があります。
- DMD遺伝子活性がほとんどない、あるいはまったくない人は、デュシェンヌ型筋ジストロフィー(DMD)を発症します。DMDは、深刻な進行性の筋萎縮性疾患です。初期の症状としては、階段の昇降困難、よちよち歩き、頻繁な転倒などであり、幼児期にこれらの症状が現れます。ほとんどの患者は10~12歳頃に車椅子に依存するようになり、20歳頃には人工呼吸器が必要になります。DMD患者の平均寿命は20~40年です。2
- 部分的なDMD遺伝子活性を持つ人は、ベッカー型筋ジストロフィー(BMD)を持つ可能性があります。BMDは、後発性骨格筋の衰弱を特徴とする疾患です。発症年齢は5歳から60歳までと大きく異なり、経過はDMDよりも緩やかで、予測が困難であり、平均余命は40代半ばです。3
- DMD病原性バリアントのキャリアである母親は、DMD関連拡張型心筋症(DCM)を発症するリスクがあります。この症状は、運動低下、拡張型表現型、うっ血性心不全を特徴とし、年齢とともに徐々に増加します。
重度のDMD表現型は、通常、読み枠を破壊してジストロフィン発現を完全に失うバリアントによって引き起こされるのに対し、軽度のBMD表現型は、通常、フレーム内変異と関連しており、異常ではあるが半機能的なタンパク質(N末端とC末端が無傷)の生成につながります。4
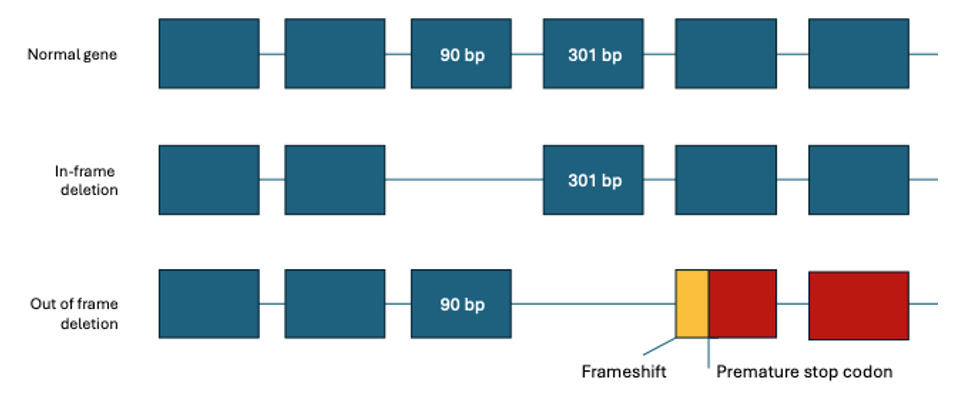
変異解析は、遺伝子サイズが大きく(2.2 MbのゲノムDNAで79エクソン)、変異タイプの多様性があるため、従来は困難でした。同定された変異のほとんどは欠失であり、DMD変異の60%~65%、BMD変異の85%を占め、約5%~10%の変異で重複が観察されています。残りの原因は、Indel、点変異、スプライシング変異などの小さな変異です。6すべての症例の約25%~33%が、de novo変異イベントに起因すると推定されています。7
デュシェンヌ型筋ジストロフィーの原因とは?
DMDのほぼすべての症例は男子に発症し、同じ家族内で複数の患者が発生する場合もありますが、その場合、母方の親戚が患者であることが条件となるため、当初、DMDはX 染色体のバリアントによって引き起こされる必要があると推定されていました。これは真実であることが判明し、1987年にLou Kunkel氏と共同研究者によって遺伝子のクローニングに成功しました。8これは、鎌状赤血球貧血のHBB(1985年)と嚢胞性線維症のCFTR(1989年)の間にある早期疾患遺伝子探索の先駆者でした。
50 bp未満の1塩基変異と小さな挿入または欠失は、確実にDMDを引き起こす可能性がありますが、DMDの病原性バリアントの80%は、1エクソン以上の欠失または重複です。9
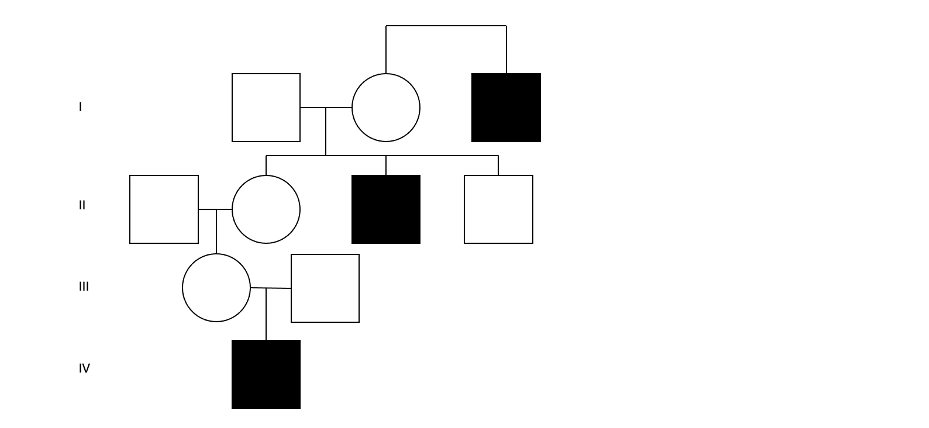
図2:DMDを発症した3人の患者を含む4世代にわたる架空の家系図。これは、X連鎖潜性遺伝の典型的なパターンを示しており、すなわち、母系を同じくする複数世代の罹患男性のパターンを示しています。
DMD治療
矯正器具や副腎皮質ステロイドなどの従来の対症療法は、プレシジョンメディシンアプローチに取って代わられつつあります。特定のエクソン挿入欠失のある患者については、アンチセンスオリゴヌクレオチド薬を使用して、特定の残りのエクソンのスキップを誘導し、DMD遺伝子の一部機能を回復させることができます。10このアプローチは、精確な分子診断に大きく依存しています。
2023年、DMDの治療薬としてより一般的な遺伝子療法が承認されました。エレビディス。本製品はウイルスベクターを使用して、DMD遺伝子の短縮版をレシピエントに導入し、自身の筋肉細胞が「マイクロジストロフィン」を生成する原因となります。この遺伝子産物のコンパクトな形態には必要な構造的および機能的コンポーネントがすべて含まれており、米国食品医薬品局は、その臨床試験のデータから、この作用が若年患者の「臨床的利益を予測する可能性が適度に高い」ことが示されると結論付けています。これにより、この疾患やその他の疾患に対する遺伝子治療アプローチが定着することを期待しています。
DMDバリアントは、通常どのように検出されますか?
マルチプレックスライゲーション依存プローブ増幅(MLPA)は、DMDに広く適用されているコピー数バリアント(CNV)を解析するための定評のある手法です。この増幅ベースのアプローチは、最も一般的な病原性DMDバリアントの検出に効果的ですが、1塩基変異(SNV)や小さなIndelを検出できないことなど、複数の主要な欠点があります。
次世代シーケンサー(NGS)の最近の進歩とバイオインフォマティクスアルゴリズムの改善により、最終的には、CNVやシーケンスバリアントを含むすべてのバリアントタイプを検出するための単一のNGSベースのアッセイが開発されました。遺伝子全体のフルシーケンス解析により、深いイントロン病原性バリアントの検出と、臨床試験の結果に影響を与える可能性のある同様のエクソンが関与するCNVの精確なブレークポイント検出が可能になります。11
記載されている標準的なアプローチでは、コーディング領域に影響を与えるバリアントのほとんどを検出できますが、逆位などのサブエキソニック、サブイントロン、ディープイントロン、または大規模な構造配列は検出されず、DMD症例の2%~7%は未解決のままです12。しかし、臨床全ゲノムシーケンスでは、SNV、CNV、および小さな構造多型(SV)などのタンパク質コーディング領域と非タンパク質コーディング領域の両方にわたってほとんどの遺伝的バリアントをキャプチャーできます。
ゲノム時代の到来は、DMDにとってどんな変化をもたらしますか?
全ゲノムシーケンス(WGS)は、ブレークポイントにまたがるリードの解析とカバレッジ深度の変化を評価する統合アプローチにより、これらの限界を解決する見込みがあります。WGSは、ハイブリッドキャプチャーまたはアンプリコンベースのシーケンスとは対照的に、ラボごとに、また被験者ごとに一貫しているため、これらのゲノム再構成イベントを検出するために開発された方法は、誰でも利用できます。これらの方法の詳細については、CNV-SV共同呼び出しに関する前回のブログ記事をご覧ください。
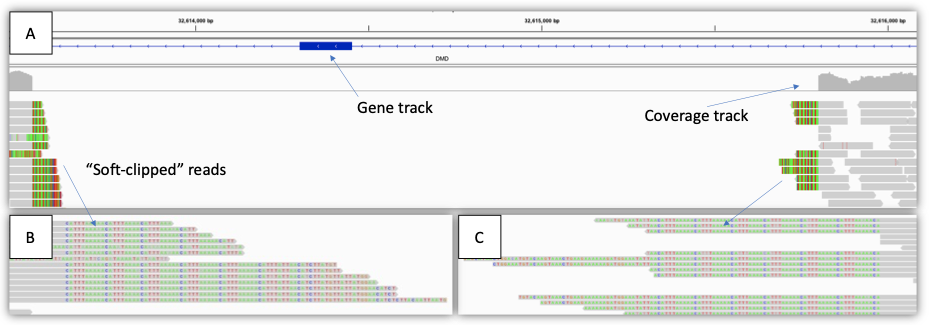
図3:デュシェンヌ型筋ジストロフィーの男性における単一エクソン欠失(2 kb)の例:(A)IGVビューは、遺伝子トラック、カバレッジトラック、アライメントされたリードトラックなど、影響を受ける領域を示しています。領域全体のカバレッジがゼロに低下し、ヘミザイガス欠失を示します。(B)、(C)のIGV拡大表示で、「soft-clip」リードを表示します。各リードの灰色のセグメントはリファレンスシーケンスと一致しますが、色付きの文字は一致しません(ソフトクリップ)。この場合、soft-clip リードは、欠失ブレークポイントのその他の端にマッピングされます。これらのリードは、SVコールの生成に使用できます。
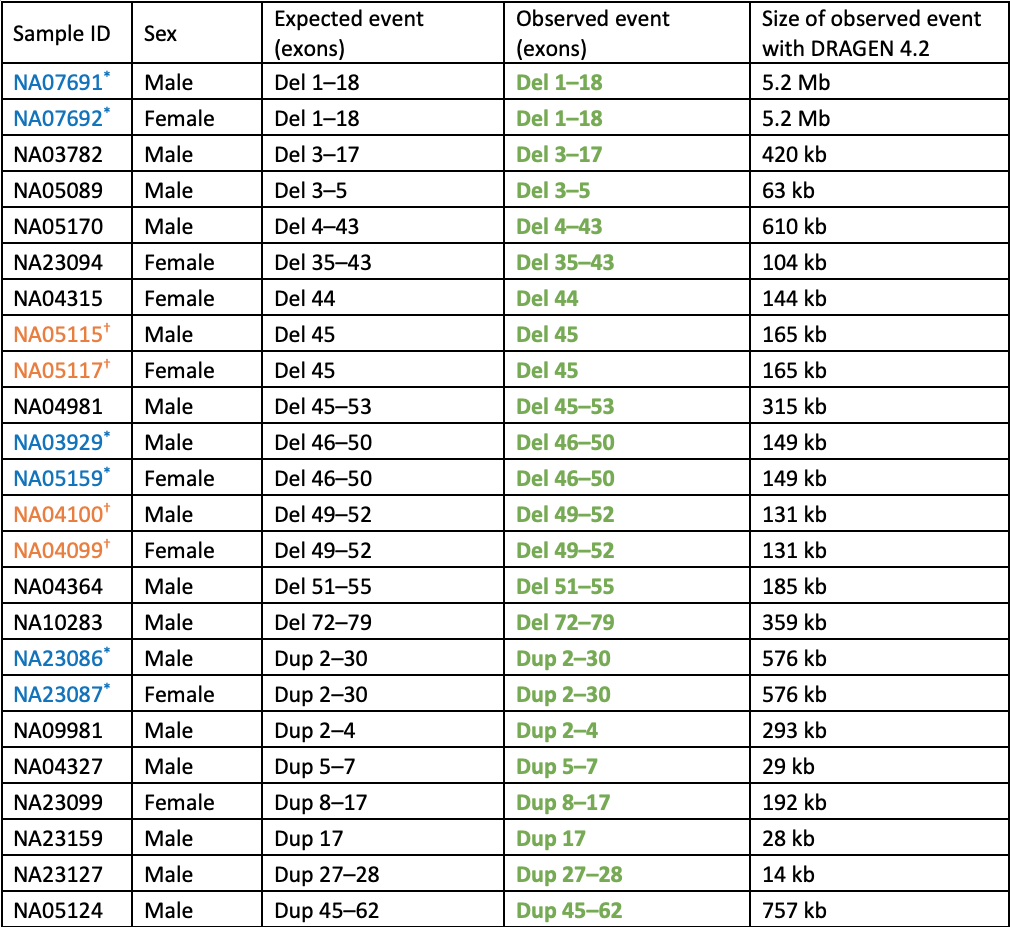
表1:WGSおよびDRAGEN 4.2 Joint SV callerによって解析された既知のDMDコピー数バリアントを持つ細胞株 — 予想されるすべてのイベントが検出されました。サンプルIDが青またはオレンジで*または†と表示されている連続入力は、男性が女性キャリアの息子である二重入力です。緑色で囲まれた太字は、予想されるエクソン、CNVタイプ(delまたはdup)、および接合状態に一致するイベントです。女性ではすべてのイベントがヘテロ接合性で、男性ではヘミ接合性です。
特に、1,000ゲノムプロジェクトデータセットの残りの約3,200人の個人について、偽陽性のエクソンDMDコピー数イベントは呼び出されませんでした。これは、集団ゲノミクスまたはキャリアスクリーニング研究などのハイスループットプロジェクトで重要な、このバリアントタイプ全体の非常に低い偽陽性率を示しています。
結論
WGSとDRAGENを使用したCNVとSVの共同コールは、極めて高感度の組み合わせであり、DMDにおける医学的に関連する欠失と重複を検出することができます。この遺伝子は、おそらく女性キャリアにおいて、コピー数バリアントコールで最も難しい遺伝子として知られているため、このエビデンスは、WGSおよびDRAGEN 4.2が非常に高い基準を設定していることを示しています。これらのアウトプットは、Emedgeneの三次解析および研究レポート作成ソフトウェアへのインプットとして互換性があり、1塩基変異やIndel、ミトコンドリアDNAバリアント、さらにはショートタンデムリピートなどの小さなゲノムバリアントに対する説明可能なAIによるバリアントの優先順位付けが含まれます。つまり、サンプルから回答までイルミナのツールを使用して、DMDのシームレスな研究評価を行うことができます。
また、これは全般化アプローチであるため、医学的に関連性のあるエクソンレベルのCNVを持つその他の遺伝子も、これらの方法を使用して評価できます。検査に同様のバリアントが重要な例としては、家族性高コレステロール血症のLDLR、神経線維腫症のNF1、遺伝性乳がんと卵巣がんのBRCA1/2、早期発症パーキンソン病のPRKNなどがあります。
学術用途向けの詳細情報またはDRAGEN試用版ライセンスについては、dragen-info@illumina.comまでお問い合わせください。
注釈
- Yoshida M, Ozawa E. Glycoprotein complex anchoring dystrophin to sarcolemma. J Biochem. 1990 Nov;108(5):748-52. doi:10.1093/oxfordjournals.jbchem.a123276
- Mercuri E, Bönnemann CG, Muntoni F. Muscular dystrophies. Lancet. 2019 Nov 30;394(10213):2025-2038. doi:10.1016/S0140-6736(19)32910-1
- Darras BT, Urion DK, Ghosh PS, et al. Dystrophinopathies. In: Adam MP, Feldman J, Mirzaa GM, et al., eds. GeneReviews. 2000 Sep 5. PMID:20301298
- Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1998 Jan;2(1):90-5. doi:10.1016/0888-7543(88)90113-9
- Le Rumeur E. Dystrophin and the two related genetic diseases, Duchenne and Becker muscular dystrophies. Bosn J Basic Med Sci. 2015 Jul 20;15(3):14-20. doi:10.17305/bjbms.2015.636
- Ji X, Zhang J, Xu Y, et al. MLPA Application in Clinical Diagnosis of DMD/BMD in Shanghai. J Clin Lab Anal. 2015;29(5):405-411. doi:10.1002/jcla.21787
- Santos R, Gonçalves A, Oliveira J, et al. New variants, challenges and pitfalls in DMD genotyping: implications in diagnosis, prognosis and therapy. J Hum Genet. 2014;59(8):454-464. doi:10.1038/jhg.2014.54
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509-517. doi:10.1016/0092-8674(87)90504-6
- Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36(4):395-402. doi:10.1002/humu.22758
- Matsuo M. Antisense Oligonucleotide-Mediated Exon-skipping Therapies: Precision Medicine Spreading from Duchenne Muscular Dystrophy. JMA J. 2021;4(3):232-240. doi:10.31662/jmaj.2021-0019
- Nallamilli BRR, Chaubey A, Valencia CA, et al. A single NGS-based assay covering the entire genomic sequence of the DMD gene facilitates diagnostic and newborn screening confirmatory testing. Hum Mutat. 2021;42(5):626-638. doi:10.1002/humu.24191
- Andrews JG, Galindo MK, Thomas S, Mathews KD, Whitehead N. DMD Gene and Dystrophinopathy Phenotypes Associated With Mutations: A Systematic Review for Clinicians. J Clin Neuromuscul Dis. 2023;24(4):171-187. doi:10.1097/CND.0000000000000436