はじめに
構造多型(SV)は、個体間のヌクレオチドシーケンスの違いの大部分を占めており、多くのヒト疾患に関与しています1, 2。しかし、特にショートリード全ゲノムシーケンス(WGS)データでは、リファレンスゲノムからの大幅な偏差によりアライメントが困難になる可能性があるため、SVの精確な検出は依然として課題となっています3。最近のロングリードシーケンステクノロジーの進歩によりSVの検出は容易になりましたが、その応用はコスト、スループットによって制限され、相当量のDNAを必要とします(イルミナのPCRフリープロトコールではゲノムあたり約25ngであるのに対して、ゲノムあたり約5ug)4。あるいは、テクノロジーの組み合わせを使用して構築できるリファレンスSVデータベースから構築されたグラフ法を使用して、既知のバリアントをジェノタイピングすることで、集団レベルでSVを研究することができます5。これらのリファレンスデータベースが進化するにつれ、集団シーケンスデータを最新の情報で再ジェノタイピングしてSVバリエーションの全体像をより詳しく把握することができます。
この記事では、ショートリードでシーケンスされた大量のサンプル集団において既知のSVをジェノタイピングできるグラフベースのジェノタイパーであるParagraphの性能を評価します。Paragraphは、有向非巡回グラフ(DAG)を構築し、その経路にリファレンスアリルとあらゆる可能な代替アリルを表現します。その後、リードをこのシーケンスグラフに再アライメントし、再アライメントされたリードに基づいて構造多型(SV)のジェノタイピングを行います(図1)。ロングリードでシーケンスされた3つのサンプルから構築されたブレークポイント精度の高いSV真理値セットにParagraphを適用しました6。真理値データの構築に用いたサンプル中のSVのジェノタイピングによる精度の評価に加え、1,000ゲノム集団内の2,501人の無関係な個人からの真理値セットSVのジェノタイピングを行い、集団遺伝学メトリクスを使用してSVコールの精度を定量しました7、15。Hardy-Weinberg平衡(HWE)などのメトリクスにより、集団におけるバリアントの全体的な性能を研究し、少数のサンプルの検査だけでは検出できない可能性のある系統的エラーを同定することができます8。1000ゲノムサンプルを用いて、集団レベルのジェノタイピング統計がコールの精度向上に役立つことを実証しました。さらに、さまざまな民族におけるこれらのSVの人口統計学的分布を定量し、機能的ゲノム要素における純化淘汰の痕跡を同定しました。
結果
SVの真理値セットの構築
Genome in a Bottle(GIAB)プロジェクトデータに含まれる3つのサンプルから開始します。NA12878(HG001)、NA24385(HG002)、およびNA24631(HG005)の3つです(「データおよび資料の入手について」を参照)。これらのサンプルについて、Pacific Bioscience(PacBio)Sequelシステムを用いてロングリードシーケンスデータを生成しました。これらのサンプルは、約11,000塩基対(bp)のHiFiリードで平均30回の深度でシーケンスされました。このデータから、PBSV9を用いてSV(50bp+)をコールしました。これらのSVをマージした後、38,709の固有の常染色体SVを同定しました。Paragraphは単一の構造多型(SV)が存在する領域での解析を前提として設計されているため、複数のSVが観察された領域(例:ブレークポイントが異なる重複または近接するSV)を除外しました。これにより、20,108のSV(9,238の欠失と10,870の挿入)が、検査および集団ジェノタイピングの目的で、当社のロングリードグラウンド真理値(LRGT)として残されました。これらのSVの詳細な説明は、こちらでご覧いただけます6。
単一サンプルのコール率と精度
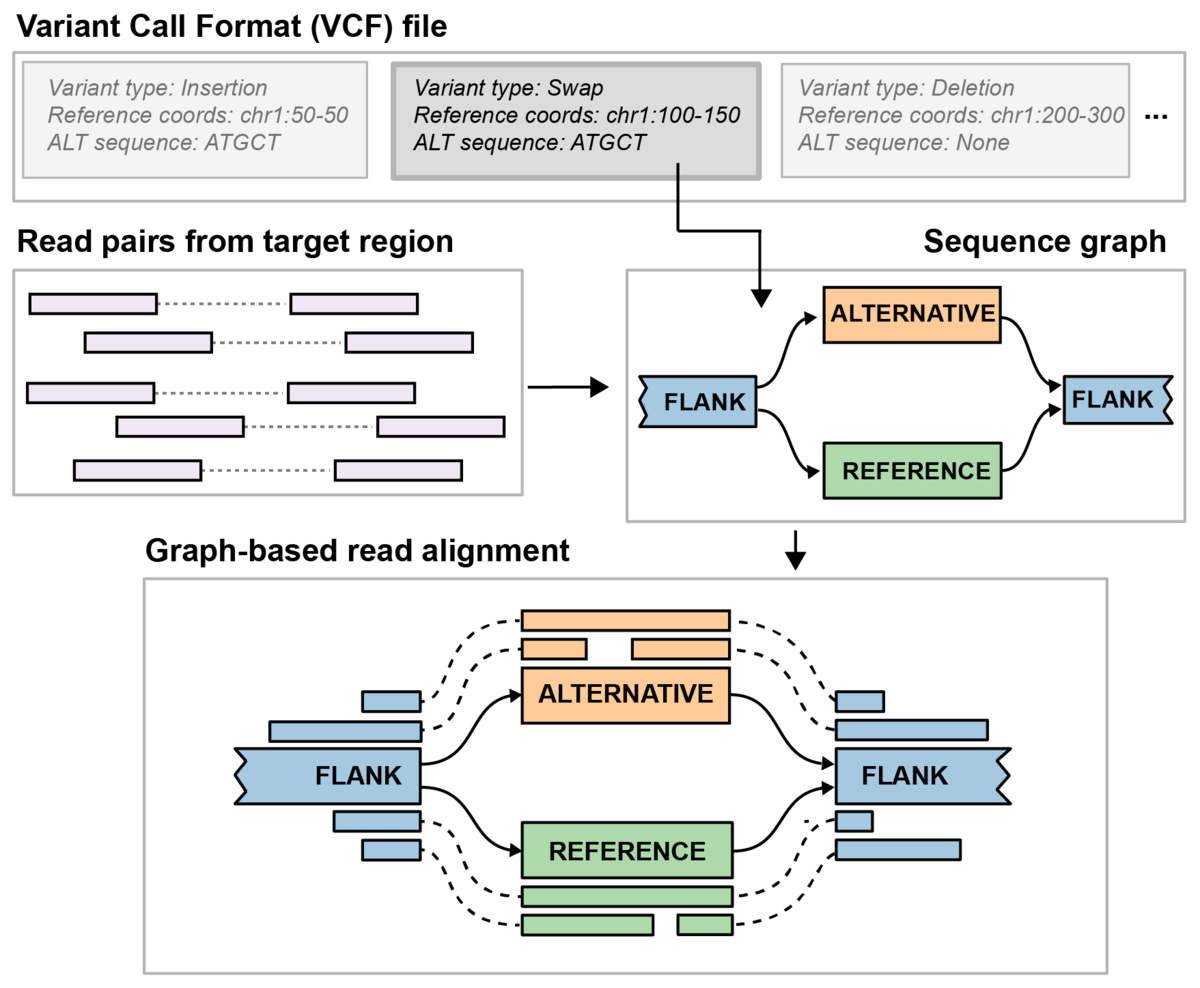
これらのSVのジェノタイピング性能を評価するため、当社のグラフジェノタイパー、Paragraphを用いてNA12878(63x)、NA24385(35x)、NA24631(40x)のショートリードデータに基づいて当社のLRGT SVのジェノタイピングを行いました(図1)6。ロングリードSVコーラーにおけるホモ接合性アリルとヘテロ接合性アリルの分類精度は系統的に評価されていないため、ジェノタイピングの一致性ではなく、バリアントの有無に重点を置いたテストを行いました。したがって、Paragraphが非リファレンスジェノタイプコールを行い、SVがLRGTにも存在する場合、バリアントを真陽性(TP)と定義し、LRGTがそのサンプルにバリアントを含まない場所でParagraphが非リファレンスアリルをコールする場合、偽陽性(FP)と定義します。この定義に基づき、LRGTには38,239の個別の代替ジェノタイプが含まれ、これが真陽性(TP)となり、22,085のリファレンスジェノタイプが偽陽性(FP)の算出に使用されます。3つのサンプルの独立したテストで平均すると、Paragraphのコール率は0.84、精度は0.88です(表1)6。
タイプ |
#真理値のTP |
コール率 |
#真理値のFP |
精度 |
Fスコア |
欠失 |
16,936 |
0.84 |
10,778 |
0.92 |
0.88 |
挿入 |
21,303 |
0.88 |
11,307 |
0.89 |
0.88 |
表1 LRGTでテストしたParagraphの性能
問題のあるコールを調べたところ、FNの59%とFPの77%が、タンデムリピート(TR)内で重複するSVで発生することが判明しました。Paragraphは、TR外のSVではTR内のSV(0.79)よりもはるかに優れたコール率(0.90)となっています。また、小規模(200bp未満)SVは、大規模(1,000bp以上)SV(約35%)よりもTR内にある可能性がかなり高い(約75%)ことも認められました。さらに、当社の原著論文では、SVブレークポイントのエラー(場所やサイズが間違っているなど)がParagraphの性能に悪影響を与えることを示しています。
2,501の多様なヒトゲノムのジェノタイピング
集団内の多くの無関係なサンプルから得られるジェノタイピングの結果により、基礎となる真理値データが存在しない場合でも、HWEなどの集団遺伝学測定でバリアントコーラーの精度を評価できます。Paragraphのジェノタイピング能力の実証として、また集団統計を研究するために、公開されている1,000ゲノムシーケンスリソースから、無関係の個人(NA24385またはNA24631を除く)2,501名を対象にLRGT SVのジェノタイピングを行いました。このデータセットは、アフリカ人660名(AFR)、アメリカ人347名(AMR)、東アジア人501名(EAS)、南アジア人489名(SAS)、ヨーロッパ人504名(EUR)の民族的に多様な集団で構成されています10。すべてのサンプルは、Illumina NovaSeqプラットフォームを使用し、150 bpのペアエンドリードで少なくとも30倍の深度でシーケンスされました。シーケンスデータはDRAGEN v3.5.7bワークフロー14で処理されました。
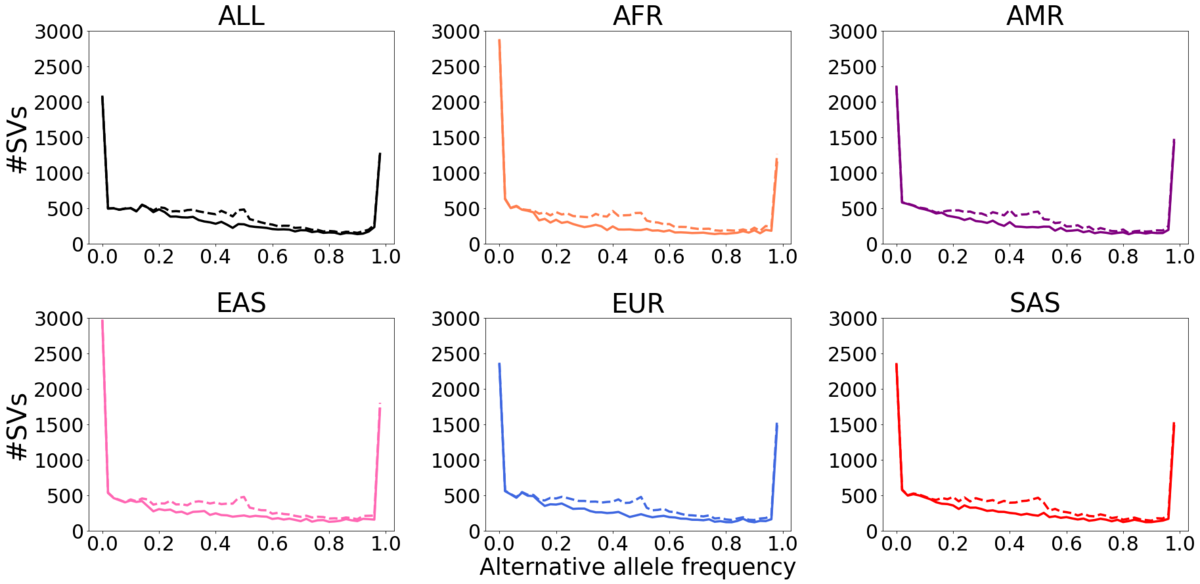
破線:すべてのSV。実線:少なくとも1つの集団でHWEテストに合格したSV。
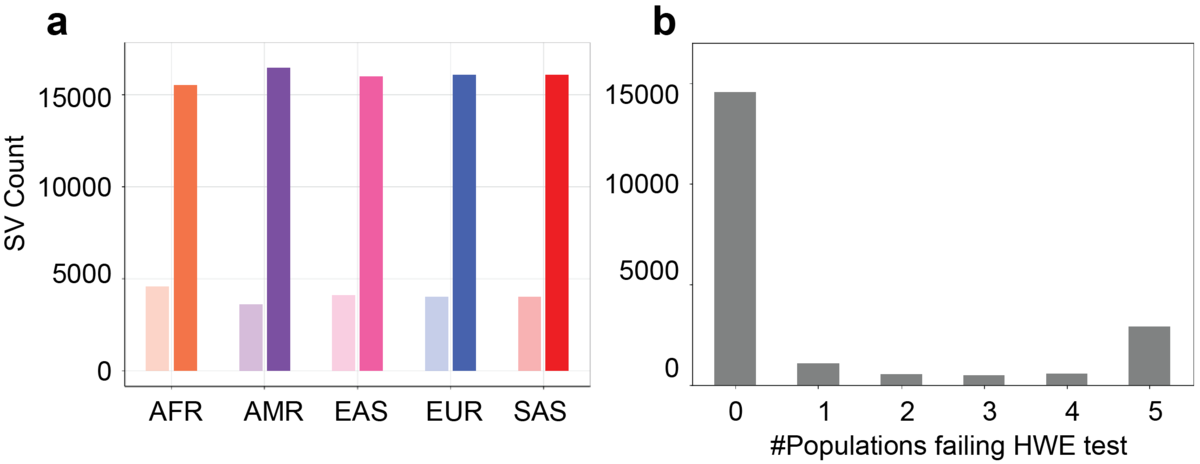
a) 各集団における、HWEに合格のSV(実線のバー)および不合格のSV(淡色のバー)のカウント。b) 少なくとも1つの集団でHWEテストに不合格となったSVについて、HWEテストに不合格となった集団のカウント。
5つの集団すべてで14,568(73%)のSVがHWEテストに合格しました。いずれかの集団でHWEテストに合格しなかったSV数は2,904(14%)でした。一部の集団ではHWEテストに合格したが、5つすべてには合格しなかったSVは2,636(13%)でした(図3b)。5つの集団すべてでHWEテストに不合格となったSVのうち、78%がTR(タンデムリピート)と重複しています。TRでは、ゲノムグラフの異なるパスは非常に似ている可能性があり、Paragraphで採用された現在のアライメントは確率的にほぼ、多数のヘテロ接合性遺伝型につながります。これは、TRと重複するSVのジェノタイピング性能を向上させるために、異なるグラフジェノタイピングモデルが必要であることを示唆しています。HWEに一致しないSVを除去した後、予期しない対立遺伝子頻度(AF)のピーク(約0.5付近)が消失することを確認しました(図2、実線)。一部の集団でHWEに一致しなかった14%のSVの大半は、極端に有意なHWE p値を示しておらず、これらのSVが一部のサンプルや特定の集団でブレークポイントのずれを持つ可能性があることを示唆しています。
混合集団におけるSV分布
HWEテストで不合格となったSVをフィルタリングした後、AFの全体的な分布はすべての集団で類似しています。このAFの類似性にもかかわらず、HWEに合格したSVの遺伝子量(ホモ接合性リファレンスジェノタイプおよび欠損ジェノタイプを0、ヘテロ接合を1、ホモ接合代替ジェノタイプを2とする)に基づく主成分分析(PCA)では、LRGT SVの集団特異性が明らかになりました(図4a)。第1および第2の主成分を2次元空間に投影すると、AFR、EAS、EURおよびSASサンプルは、明確に分離されています。逆に、AMRサンプルは、混合的な集団に予想されたとおりに、さまざまな祖先集団間でマッピングされます。
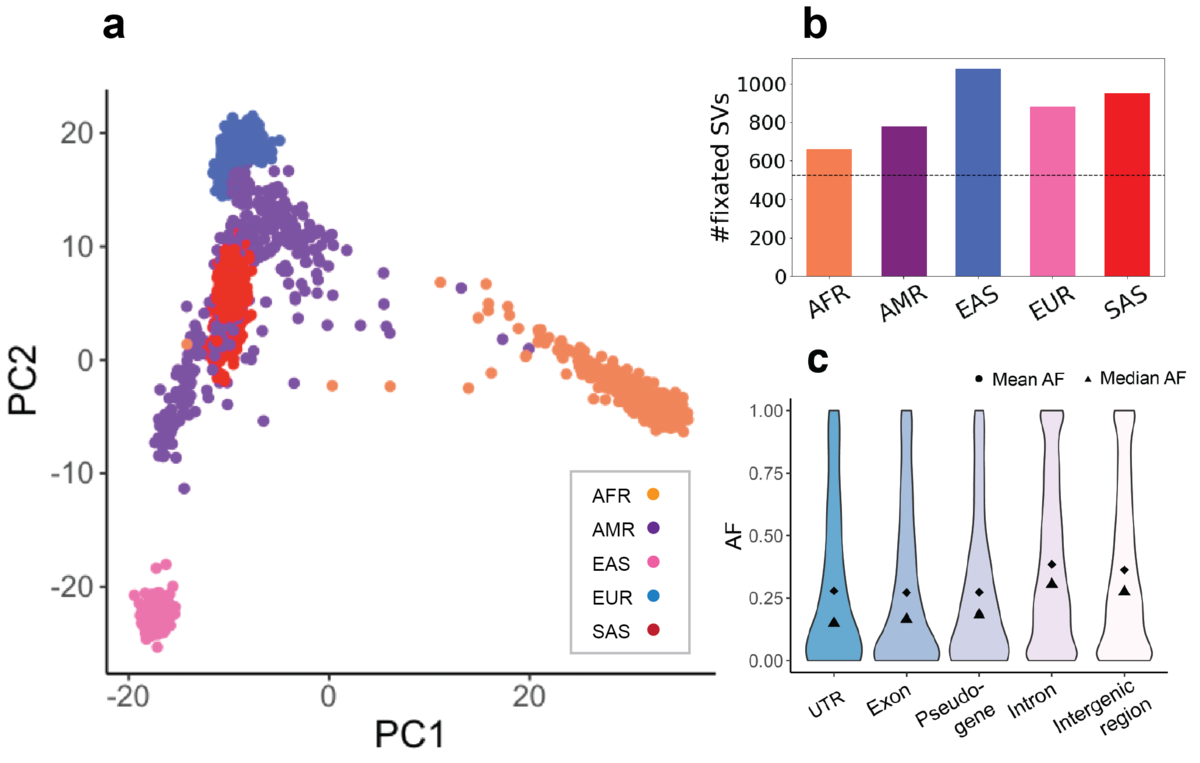
a)HWE合格SVの1000ゲノムサンプルのPCAバイプロット分析。b)各集団における固定SV数。c)全集団にわたる異なる機能要素におけるSV AF。
5つの集団すべてで固定された合計で525のSVが観察されており、リファレンスゲノムの希少なアリルを表している可能性があります(図4b)。これらの他に、少なくとも1つの集団に固定された715のSVがあります。AFRは、最も分散した集団であることと一致する形で、他の集団よりも固定SVが少なくなっています。機能的アノテーションと組み合わせると、303の固定SVはエクソン性です。UBE2QL1における1,638 bpのエクソン挿入も、過去の2件の研究で非常に高頻度に報告されました12, 13。特に、TOPMedによる最近の研究では、混合祖先からシーケンスされた53,581人のすべての人でこの挿入が報告されています13。
また、ゲノム状況に基づくSVの別のAFも観察されました。一般的に、コード配列のエクソン、偽遺伝子、および非翻訳領域(UTR)内のSVは、イントロン領域および遺伝子間領域よりもアリル頻度が低くなります。イントロンおよび遺伝子間領域のSVは、機能性要素(UTR、エクソン)のより極端なAFと比較して、より均一なAF分布を示します(図4c)。これらはすべて、機能的な結果をもたらす可能性のあるSVに対する純化淘汰を示唆しています7。一般的なSVは機能的要素において少ない傾向がありますが、TP73(集団全体のAF=0.16、がん抑制遺伝子)、FAM110D(集団全体のAF=0.65、機能未解明、細胞周期に関連する可能性)、OVGP1(集団全体のAF=0.20、受精と早期胚発生に関連)など、遺伝子のエクソン内にいくつかの一般的なSVが見られます。具体的には、TP73の欠失はサブ集団で頻度が大きく異なります。AFRでは0.02と低く、EURとEASでは0.20より高いです。
結論
この記事では、GIABコンソーシアムに含まれる3つのサンプル用に作成したSVの真理値セットの初版を簡単にレビューしました。この真理値セットは、グラフ手法により高品質のSVのデータベースのジェノタイピングが可能となることを示しています。当社の継続的な取り組みにより、このデータベースが拡張され、Paragraphが改善されれば、より精確なジェノタイピングが可能となります。多様性のある1000ゲノム集団でこれらのSVをジェノタイピングすることで、単一サンプルに基づく真理値データがない場合でも集団データを品質管理とし使用できる方法を提示しました。さらに私たちは、基礎となる真理値データで同定されたエクソンSVの多くが1000ゲノムサンプルでも検出され、淘汰の兆候が示されていることを実証しました。Paragraphを使用してWGSのコホートでこれらのエクソンSVを直接ジェノタイピングすることで、これらのSV/遺伝子の生物学についての理解をさらに深めることに役立つでしょう。
学術用途向けの詳細情報またはDRAGEN試用版ライセンスについては、dragen-info@illumina.comまでお問い合わせください。
データおよび資料の入手について
Paragraphソフトウェアは、https://github.com/Illumina/paragraphから一般に利用可能です。
NA12878, NA24385、およびNA24631 PacBioデータは、PRJNA540705、PRJNA529679およびPRJNA540706としてSRAに保管されています。イルミナのデータは、PRJEB35491としてENAに保管されています。
Dragenが再処理した1000ゲノムデータは、https://registry.opendata.aws/ilmn-dragen-1kgp/から入手できます。
注釈
- Weischenfeldt, J., Symmons, O., Spitz, F. & Korbel, J.O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat Rev Genet 14,125-138 (2013).
- Lee, C. & Scherer, S.W. The clinical context of copy number variation in the human genome. Expert Rev Mol Med 12, e8(2010)。
- Goodwin, S., McPherson, J.D. & McCombie, W.R. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17, 333-351 (2016).
- Ashley, E.A. Towards precision medicine. Nat Rev Genet 17, 507-522 (2016).
- Logsdon, G.A., Vollger, M.R. & Eichler, E.E. Long-read human genome sequencing and its applications. Genome Biol 20, 291 (2019).
- Chen, S. et al. Paragraph: a graph-based structural variant genotyper for short-read sequence data. Genome Biol 20, 291 (2019).
- Sudmant, P.H. et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75-81 (2015).
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987-2993 (2011).
- Wenger, A.M. et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol 37, 1155-1162 (2019).
- Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68-74 (2015).
- Wigginton, J.E., Cutler, D.J. & Abecasis, G.R. A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet 76, 887-893 (2005).
- Sherman, R.M. et al. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat Genet 51, 30-35 (2019).
- Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. bioRxiv, 563866 (2019).
- Chu E, Friedman A. DRAGEN reanalysis of the 1000 Genomes Dataset now available on the Registry of Open Data. Amazon Web Services, 2020.
- Bryska-Bishop, M. et al. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. bioRxiv, 430068(2021)。