NovaSeq Xシリーズの出荷開始により、イルミナの次世代ハイスループットプラットフォームでXLEAP-SBSケミストリーが利用可能になりました。さらに25Bフローセルが利用可能になり、NovaSeq Xソフトウェアアップデート1.2は、デュアルフローセルシーケンスランで最大16テラ塩基を生成できるようになりました。このアップデートにより、NovaSeq X Plusは年間数万の全ゲノムを生成できます。1.2のリリースでは、より高いスループットに加え、ワークフローがアップグレードされ、精度が向上し、Qスコアのキャリブレーションが改善されています。改善されたQスコア精度とXLEAP-SBSケミストリーの組み合わせにより、NovaSeq Xの中品質および高品質のbinは、それぞれQ20からQ24、Q37からQ40にシフトします。装置のより高度でより正確な塩基の品質は、XLEAP-SBSケミストリーとNovaSeq Xシリーズプラットフォームの性能をより良く反映しています。
この記事では、以下の点に重点を置いています。
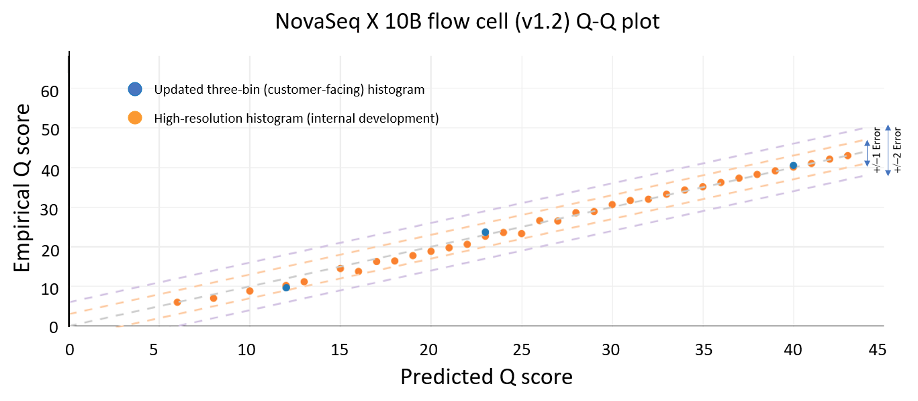
- シーケンスおよびキャリブレーションワークフローの改善により、NovaSeq Xが、経験的精度との優れた相関性で、85%の塩基を最高品質のbin(Q40)で出力することが可能になる
- QスコアのキャリブレーションプロセスでPCRフリーのライブラリー調製を使用することにより、サンプル調製の影響を排除し、シーケンスの品質をより正確にQスコアに反映することができる
- Qスコアを3つのbinに量子化することによるデータ管理とコスト面の利点
- Qスコア量子化が下流のアプリケーションに与える影響
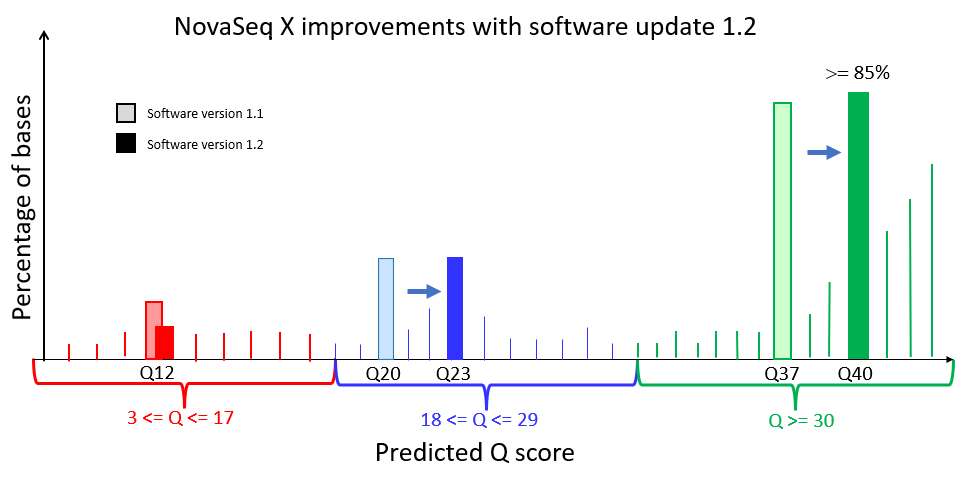
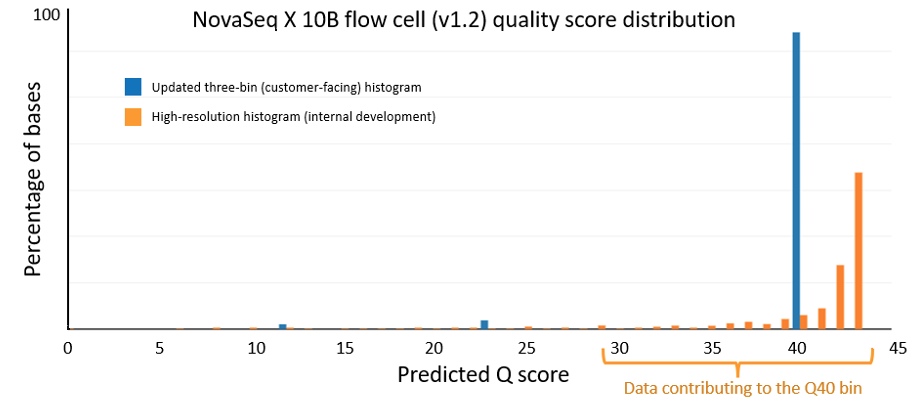
下の図1は、NovaSeq X 1.2アップデートにおける改善を示すQスコアヒストグラムです。図の細い線は、特定の整数値のクオリティスコアを持つ塩基の割合を表します。NovaSeq Xは、お客様のコストの削減とデータ転送時間の短縮のために、これらの整数間隔のクオリティスコアを、Q12, Q24、Q40のラベルが付いた濃いバーで示される3つのbinに集約します。これらのbinには、クオリティスコアがそれぞれ3~17、18~29、30以上の範囲の塩基が含まれています。各濃い色のバーの下のラベルは、その集計されたbin内のすべての塩基のクオリティスコアを表します。ラベルはbinされたデータの平均化した品質を表しており、binはラベル化された値を上回るもしくは下回る、個々の高解像度のクオリティスコアを持つ塩基を自然に集約します。Qスコアは対数スケールであるため(以下の詳細を参照)、分布は非対称で、集約されたbinは主に高品質の塩基で構成されています。1.2へのアップデートにより、より正確な表現とクオリティスコアの上方へのシフトがもたらされ、最も高い品質の塩基のスコアがQ40とレポートされるようになります。NovaSeq Xは、出力塩基の>= 85%をQ40のスコアで生成することができます。また、これらのQ40スコアの塩基のほとんどが、高解像度のQスコアでもQ40を上回っています。
Qスコアの基礎:ベースのクオリティスコアとは何ですか? Phredスコアとは何ですか?
各塩基がイルミナシーケンサーでデコードされると、ベースコーリングモデルは、割り当てられたベースコールが正しい確度を表す信頼度スコアを割り当てます。1 ベースコールエラーのraw probabilityは、電力測定に使用されるdB命名法のように、ダイナミックレンジを圧縮するために対数スケールに変換されます。結果として得られるスケーリング後の値は、塩基のクオリティスコア(またはQスコア、またはPhredスコア)として知られています。例えば、ベースコーラーがベースコールの信頼度が99%である場合、エラーの確度は1%で、Phred(Q)スコアは20になります。
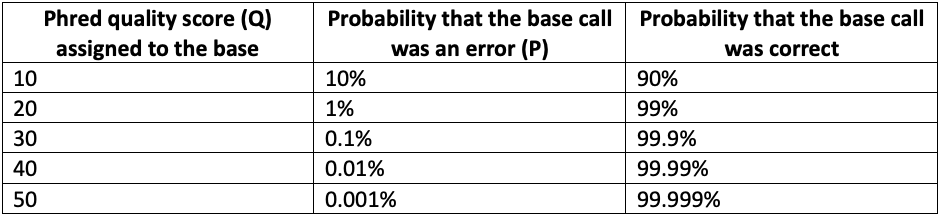
数学的には、PhredクオリティスコアはQ = -10 log10 Pと定義され、Pはベースコーリングエラーの確度です。Phredスコアが増加すると、以下の表1に示すように、ベースコールの信頼度は非線形的に増加します。
Qスコアエラーの原因:すべてのエラーがシーケンサーで生成されるわけではありません
適切にキャリブレーションされたQスコアモデルにより、シーケンスエラーの確度が予測されます。ただし、ノイズや低シグナル強度など、シーケンスプロセス自体に起因するエラーもありますが、その他のエラーはシーケンサーの上流から発生することを理解することが重要です。NGSワークフローにおける一般的なエラーの原因を以下の図5に示します。
シーケンスの前に、サンプル抽出とライブラリー調製により、ゲノムDNAサンプルを断片化ライブラリーに変換します。このプロセスは通常、断片化、各断片の両端への専用アダプターの追加、増幅または精製ステップを伴います。いくつかの論文では、これらのプロセスによって導入されうるエラーについて説明しています。2-4 シーケンサーは、サンプル調製によって導入されたエラーを持つ塩基と、サンプル内の他の塩基とを区別することはできません。サンプル調製エラーは、上流で静かに導入されます。したがって、サンプル調製エラーの推定と軽減は、エラー補正アルゴリズムを通じて下流パイプラインで実施する必要があります。

クオリティスコアキャリブレーションのトレーニングに使用される手法には、サンプル調製とシーケンスプロセス自体の両方からのエラーが自然に含まれています(付録を参照)。PCRフリーのデータのみを使用するキャリブレーションでは、PCRフリーライブラリー調製によりサンプル調製によって導入されるエラーが最小限に抑えられるため、実際のシーケンサーのベースコーリング精度をより正確に反映するクオリティスコアが得られます。
下流の解析の再キャリブレーションプロセスでは、新しいデータセットでシーケンスおよびサンプル調製エラーを動的に特性化し、高感度アプリケーションの精度を向上させることができます。さらに、これらのキャリブレーションプロセスは、特定のエンドユーザーパイプラインに合わせてカスタマイズでき、サンプル調製エラーのシグネチャのバリエーションを補います。Broad InstituteのGenome Analysis Toolkit(GATK)のベストプラクティスには、割り当てられた塩基の品質と経験的塩基の品質の間のマッピングを推定する塩基のクオリティスコア再校正(BQSR)ステップが含まれます。5BQSRの難しさは、既知のバリアントサイトのデータベースが必要な点であり、ほとんどのサンプルでこれを利用することはできません。
DRAGENパイプラインには、パイプライン/サンプル塩基ごとにエラーメカニズムを推定する高度なアルゴリズム6が含まれており、truthセットへのアクセスを必要とせず、PCR関連アーティファクトに特に重点を置いています。DRAGENはBQSRを必要とせず、使用もしません。
Qスコア解像度:膨大なゲノムデータがベースクオリティのラベリングの最適化を促進し、保管容量を低減
イルミナのシーケンサーのデータスループットが時間とともに大幅に増加するにつれ、イルミナは、お客様のデータ転送時間と保管コストを削減するために量子化されたクオリティスコアを採用しました。
MiSeqシステムなどのイルミナの初期の装置は、50の整数binを使用してQスコアを生成しました。シーケンサーの出力データのストレージフットプリントを大幅に削減するために、新しいシステムでは、Qスコアの連続間隔をより少ないbinで表します。このアプローチは、クオリティスコアのエントロピーを減らし、より高いレベルのデータ圧縮を可能にします。NovaSeq 6000、NextSeq 1000、NextSeq 2000、iSeqはすべて3 bin Qスコアテーブルを使用します。図6は、クオリティスコアを少ないbin数に集約する利点を示しています。FASTQファイル(gzip形式)とBAMファイルのストレージサイズは50%も削減され、ストレージコストの節約に直接つながります。
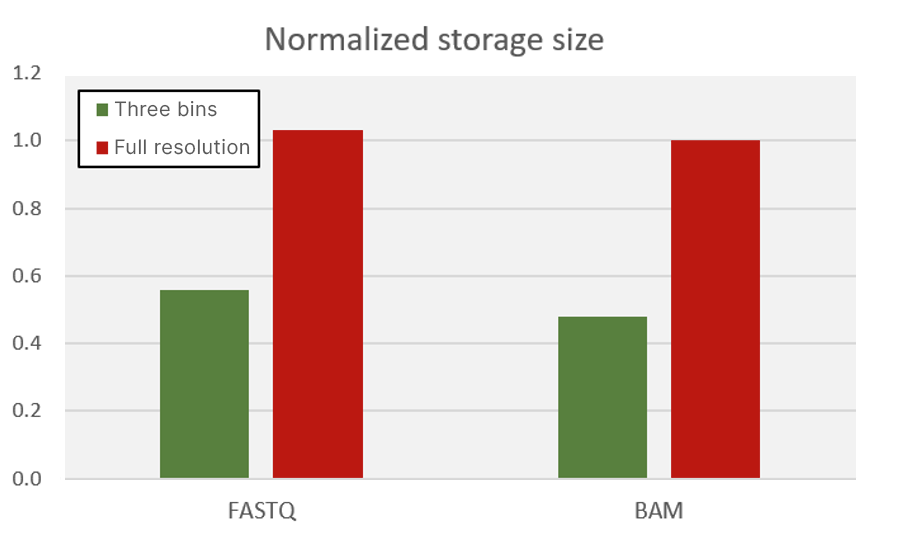
llluminaとそのユーザーは、Qスコアの解像度の低下が下流のパイプラインにほとんどまたは全く影響しないことを示し、データ転送時間を大幅に短縮し(大規模なシーケンスプロジェクトや生産規模のシーケンスでは非常に重要)、同時に必要なデータストレージを大幅に削減する(ユーザーのコスト管理において非常に重要)ことを実証しています。詳細については、このトピックに関するイルミナのアプリノートをご覧ください。7
下流のアプリケーションにおけるQスコアの重要性:Qスコアは重要ですか?
Q35を超えるクオリティスコアは、最先端のバリアントコーラーを使用する生殖細胞系列アプリケーションに与える効果は限定的です。精度は、ベースのクオリティスコアではなくカバレッジによって大きく制限されます。カバレッジ要件はポアソンサンプリングが中心です。例えば、Journal of Computational Biologyのこの論文を参照してください。8 バリアントのアリル頻度がはるかに低い、高感度の体細胞アプリケーション(微小残存病変を含む)が開発されているため、Qスコアが高いほど必要な生のシーケンスデータの量を減らすことができます。DRAGEN体細胞パイプラインには、サンプル調製中に生じたエラーの検出を改善するアルゴリズムが含まれています。9
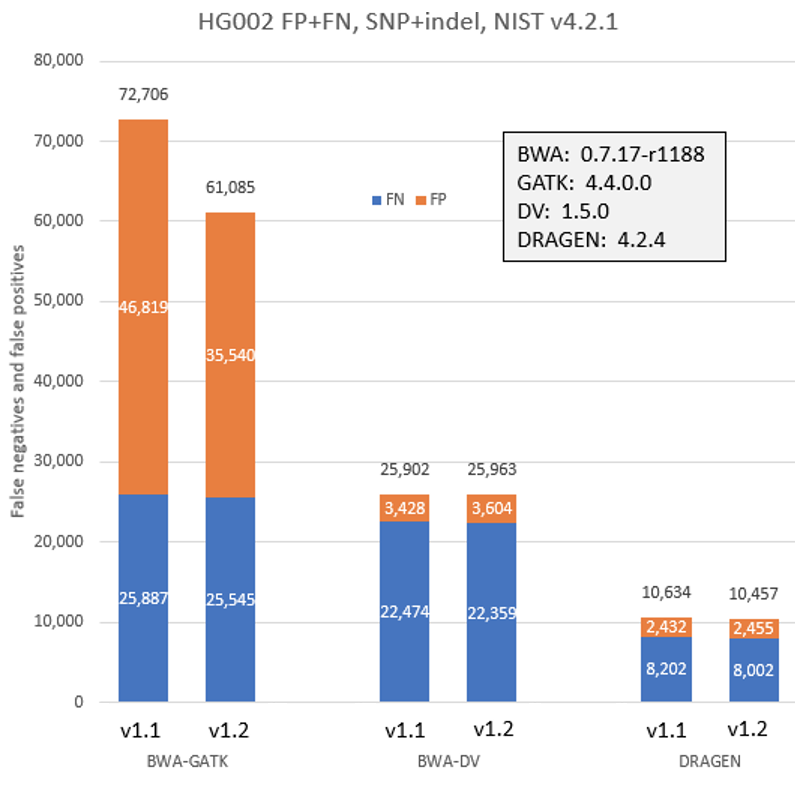
1.2アップデートからの新しいワークフローとQスコア予測の改善が二次解析に与える影響を確認するために、バージョン1.1と1.2の3つの定評のある解析パイプラインでHG002ゲノムサンプルを解析しました。以下の図7に示すように、GATKの精度結果はQスコアの改善とともに改善しました。DRAGENおよびDeepVariantのパフォーマンスは、Qスコアの改善とともに有意な変化を示しませんでした。
結論
NovaSeq Xソフトウェア1.2のアップデートのリリースは、XLEAP-SBSケミストリーとNovaSeq Xプラットフォームのシーケンス品質をより正確に反映しています。NovaSeq Xは、より正確なベースコーリング、クオリティスコアリング、binningにより、最高(Q40)のbinに> 85%の塩基を供給できるようになりました。
さらに、NovaSeq Xソフトウェアの1.2バージョンには、クオリティスコアを割り当てる際に以下の重要な考慮事項が組み込まれています。
- 塩基のクオリティスコアは、統計的なシーケンサーエラー率を捉え、基準となるグラウンドトゥルースなしでシーケンス上で推定されます
- サンプル調製プロセスで生じるエラーを最小限に抑えるため、PCRフリーライブラリー調製法をイルミナのキャリブレーションプロセスで使用します
- 効率的なQスコアbinningは、顧客のコストとデータ転送時間を削減するために重要です
- binningは、最先端のバリアントコーラーによる生殖系列アプリケーションにほとんどまたはまったく影響しません
NovaSeq X Software 1.2のアップデートの詳細については、ここをクリックしてください。
付録 Qスコアキャリブレーショントレーニング
顧客リリース前に、シーケンサープラットフォームはキャリブレーショントレーニングを使用して、塩基のクオリティスコアの予測を最適化します。これにより、シーケンスプラットフォームで割り当てられた塩基のクオリティスコアが、経験的ベースコーリングの精度と厳密に一致するようになります。
ベースのクオリティスコアのキャリブレーショントレーニングアルゴリズムの多くは、1998年に発表された基礎的な論文から派生しており、クオリティスコアを割り当てる計算効率の高い方法について説明しています。10 ルックアップテーブルの作成には、Signal to Noise比(SN比)などの少数の予測変数を使用します。各rowは、予測因子空間の小さな領域における経験的品質を記録します。量子化された一連の予測値から塩基のクオリティスコアを簡単に生成できます。この論文では、クロマトグラムファイルから導出された4つのヒューリスティック予測因子を使用していますが、一般的なアプローチは、あらゆるタイプのシーケンスシステムにおいて、任意の数の予測因子を用いて適用できます。
経験的品質は、予測空間の所定の領域について、呼び出された塩基の総数の比率として正しく呼び出された塩基の数と定義されます。この計算に使用される塩基は、例えば、ヒトゲノムの大部分をカバーするヒトデータ用のGenome in a Bottle(GIAB)真理値セットなど、真理値が分かっている塩基に限定されます。K-merが可能な限りトレーニングで公正に表現されるように、独自の高品質の真理値セットを持つその他のゲノム(細菌、微生物)が含まれます。マッピングはシーケンス操作の一部ではなく、シーケンスエラー率の測定には寄与しないため、マッピングエラーが発生しやすい領域は計算から除外されます。
参考文献
- Illumina. Understanding Illumina Quality Scores. illumina.com/content/dam/illumina-marketing/documents/products/technotes/technote_understanding_quality_scores.pdf. 2014年発行。2023年10月9日にアクセス。
- Chen G, Mosier S, Gocke C, Lin M-T, Eshleman J. Cytosine Deamination is a Major Cause of Baseline Noise in Next-Generation Sequencing. Mol Diagn Ther. 2014 Oct; 18(5): 587–593. doi:10.1007/s40291-014-0115-2
- Otsubo Y, Matsumura S, Ikeda N, Yamane M. Single-strand specific nuclease enhances accuracy of error-corrected sequencing and improves rare mutation-detection sensitivity. Arch Toxicol. 96, 377–386 (2022). doi:10.1007/s00204-021-03185-y
- Gregory T, Ngankeu A, Orwick S, et al. Characterization and mitigation of fragmentation enxyme-induced dual stranded artifacts. NAR Genom Bioinform 2020 Dec; 2(4). doi: 10.1093/nargab/lqaa070
- Caetano-Anolles D. Base Quality Score Recalibration (BQSR) – GATK. Broad Institute Genome Analysis Toolkit. gatk.broadinstitute.org/hc/en-us/articles/360035890531-Base-Quality-Score-Recalibration-BQSR-. 2023年発行。2023年10月9日にアクセス。
- https://science-docs.illumina.com/documents/Informatics/dragen-v3-accuracy-appnote-html-970-2019-006/Content/Source/Informatics/Dragen/dragen-v3-accuracy-appnote-970-2019-006/dragen-v3-accuracy-appnote-970-2019-006.html
- Illumina. NovaSeq™ 6000 System Quality Scores and RTA3 Software. illumina.com/content/dam/illumina-marketing/documents/products/appnotes/novaseq-hiseq-q30-app-note-770-2017-010.pdf. 2017年発行。2023年10月9日にアクセス。
- Deng C, Daley T, Calabrese P, Ren J, Smith A. Predicting the Number of Bases to Attain Sufficient Coverage in High-Throughput Sequencing Experiments. J Comput Biol. 2020 Jul; 27(7): 1130–1143. doi: 10.1089/cmb.2019.0264
- Scheffler K, Catreux S, O’Connell T, et al. Somatic small-variant calling methods in Illumina DRAGEN™ Secondary Analysis. bioRXiv 2023.03.23.534011. doi:10.1101/2023.03.23.534011
- Ewing B, Hillier L, Wendl M, Green P. Base-Calling of Automated Sequencer Traces Using Phred. I. Accuracy Assessment. Genome Res. 1998;8:175–187. doi:10.1101/gr.8.3.175