はじめに
臨床試験として全ゲノムシーケンスを実装するラボが増えるにつれ、エンドツーエンドなシーケンスパイプラインの性能を精確に定量することがますます重要になっています。1つの選択肢として、リファレンス材料を取得してシーケンスを行い、それを用いて関連する真理値データと比較しながらバリアントコールの性能を評価する方法があります1-3。これにより、ゲノムの大部分(80~90%)において、コール率と精度を精確に測定することができます。しかし、これらのリファレンス材料は、限られた数の民族的に均質なサンプルに対してのみ存在し、将来のサンプルにおけるゲノムワイドの性能を必ずしも予測できるわけではありません。補完的なアプローチとして、あらゆるサンプルのシーケンス性能を推定する予測手法を開発することが挙げられます。
バリアントコールは、確率的変動と系統的変動の両方の影響を受ける可能性があります。1塩基変異(SNV)や短いIndelのような小規模バリアントに関しては、30×以上のカバレッジでシーケンスされたゲノムの大部分において、確率的な変動がバリアントコール性能に与える影響はごくわずかです。しかし、エラー率の上昇、マッピング品質の低下、深度の異常など、低品質によって系統的に影響を受けるゲノム領域では、SNVやIndelであっても、一貫して精確なバリアントコールを行えない場合があります。これらの系統的エラーの原因となるリファレンス特性の多くはよく知られています。例えば、反復性の高い領域はマッピング品質が低く、ホモポリマーは塩基精度が低いことが知られています。この知識は、容易領域と困難領域にゲノムを分類するために使用されます4。これらの分類は役に立つかもしれませんが、これらの領域における実際の性能を完全に表しているわけではありません。例えば、大きなセグメント重複は、高い類似性と低い類似性の領域で構成され、バリアントコーリング精度は大きく異なる場合があります。これらの一般的な分類方法を改善する方法は、実際のシーケンスデータを使用して、バリアントコーリング性能の高い領域と低い領域を経験的に同定することです。
ここでは、50×を超える平均深度にシーケンスされた29サンプルから収集されたデータ品質に関連するいくつかのシーケンシング指標を解析しました。これらを統合すると、ゲノムの各塩基を平均1,450リードでカバーすることが可能となり、塩基レベルの分解能で一貫して高品質な領域を同定できます。また、各座位の系統的品質を経験的に高いまたは低いとしてアノテーションすることができます。次に、これらのアノテーションは、ペアワイズSNVの一致率によって測定される、SNV性能を高度に予測できることを示します。これらの経験的に導出されたアノテーションにより、頻繁に使用されるリファレンスベースの分類法を改善することができます。例えば、Genome in a Bottle(GIAB)の困難領域内にありながら、信頼度が高いとアノテーションされた領域では、テクニカルレプリケート間で高いSNV一致率(Jaccardインデックス=98.8%)が得られています。逆に、GIABの困難領域に含まれていないが、信頼度が低いとアノテーションされた領域は、SNVの一致率が低く(Jaccardインデックス=79.9%)なっています。当社の結果は、多くのサンプルからシーケンスデータを集計することで、小規模バリアントコーリングが系統的に高い品質を持つゲノム領域を同定できることを示しています。
手法
データベースを構築するために、1000 Genomes Projectに含まれ、Coriell Institute Biobank5から取得した29のサンプルを選択しました。これらのサンプルは多様な民族(アフリカ11、東アジア9、ヨーロッパ9のサンプル)から構成されます。各サンプルからのDNAは、TruSeq™ PCR-Free Sample Prep Kitを使用して調製した後、Xpワークフローを備えたNovaSeq™ 6000装置上でペアエンドの150bpリードでシーケンスされ、平均深度は51xでした。
シーケンスリードは、デコイ染色体を含むが代替コンティグを含まないGRCh38に対して、DRAGEN™ v3.4.5を使用してアラインメントされました。サンプルが29個あるため、ゲノムの位置あたり約1,450リードが予想されます。ゲノム内の各位置のすべてのサンプルについて、アライメントファイル(BAMまたはCRAM)から、以下を含むいくつかのメトリクスを収集しました。
- 正規化深度:各サンプルについて、正規化深度は、各座位のカバレッジを常染色体のカバレッジの中央値で正規化して算出されます。そのため、値が1の場合は、常染色体のカバレッジ深度の中央値に相当します。正規化深度は、最終的なコホート深度についてすべてのサンプルで平均化されます。
- 平均マッピング品質:ゲノム位置と重複するすべてのサンプルにわたってアライメントされたすべてのリードの平均マッピング品質。
- Q20ベースコール率:全サンプルにおけるゲノム位置で、Phredスケールのクオリティスコアが20を超えるベースコールの割合です。Q20はエラー率1%以下に相当します。
この解析では、Jaccardインデックスを用いて算出された一致率を用いてSNV性能を測定します。
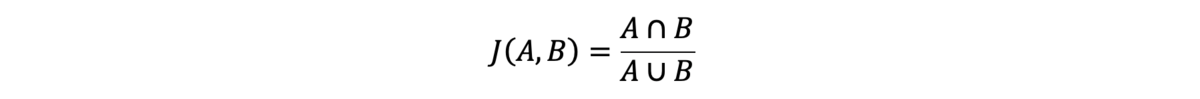
AとBは、2つのバリアントコーリングファイル(VCF)からの2セットのバリアントコールで、通常は同じサンプルをレプリケートします。分子では、AとBの共通部分は両方のVCFに存在し、同一の遺伝型を持つ、すべてのバリアントコールとして定義されます。分母では、AとBの和集合は、いずれかのVCFに存在するすべてのバリアントコールとして定義されます。これらのバリアントコールセットのJaccardインデックスは、セットからのコールの共通部分をコールの和集合で割ったものです。
基礎となる真理値リソースと比較して、一部の再現性のあるバリアントは系統的エラーが原因である可能性がありますが、結果は依然として全般的な性能評価にとって非常に有用です。すべての解析について、NA12878のランダムに組み合わせた30のテクニカルレプリケートの平均Jaccardインデックスを示します。報告された各クラスの一致率の値とバリアント数は、30のレプリケートすべての平均です。
常染色体全体の性能を計算する際、リファレンスゲノムのNとギャップを除外します。
結果
集約されたデータを使用することで、単一のシーケンシング実験では検出が困難な系統的エラーの傾向を高い精度で識別できます。ゲノム内の40リードの座位で、常染色体カバレッジの中央値が30倍であると仮定します。30×のカバレッジがポアソン分布に従うと仮定すると、ポアソンサンプリングの影響により、約1,000の独立した座位ごとに14座位の割合でこの現象が発生します。したがって、より高いカバレッジが確率的か系統的かを判断することは困難です。逆に、30×のカバレッジを持つ30人の集団で同じ座位を観察する場合、全員が座位に正確に40×のカバレッジを持っていると、座位におけるカバレッジの変動が確率的プロセスによって推進されていないことが非常に確実になります(p=2e-56)。多くのサンプルから得られる高深度データを集計することで、バリアントコール性能の低下につながる可能性が最も高い系統的異常を同定することができます。多様な民族的背景から採取した大量のサンプルでは、一般的な変異やアリル固有のアーチファクトを同定でき、それらが十分に希少であれば、集計された要約メトリクスへの影響は限定的になります。
メトリクス性能によるゲノムの層別化
手法に概説された手順に従って、3つの性能メトリクスを収集しました(図1)。
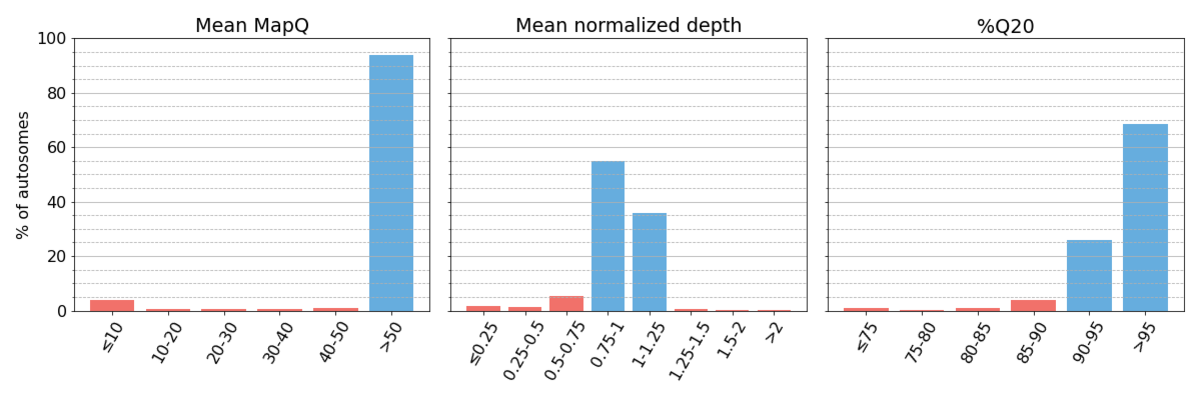
図1:集計されたシーケンスデータでゲノム全体の系統的アライメントメトリクスをbin分割しました。メトリクスbinは半開区間です。高い系統的品質を表すbinは青色で網掛けされ、低い系統的品質を表すbinは赤色で網掛けされます。これは、本稿全体を通してすべての図で維持されます。本文で説明したように、対象範囲の90%以上がQ20、平均マッピング品質が50以上、平均正規化深度がターゲットカバレッジの±25%以内の場合、高い系統的品質を持つ領域と定義しています。
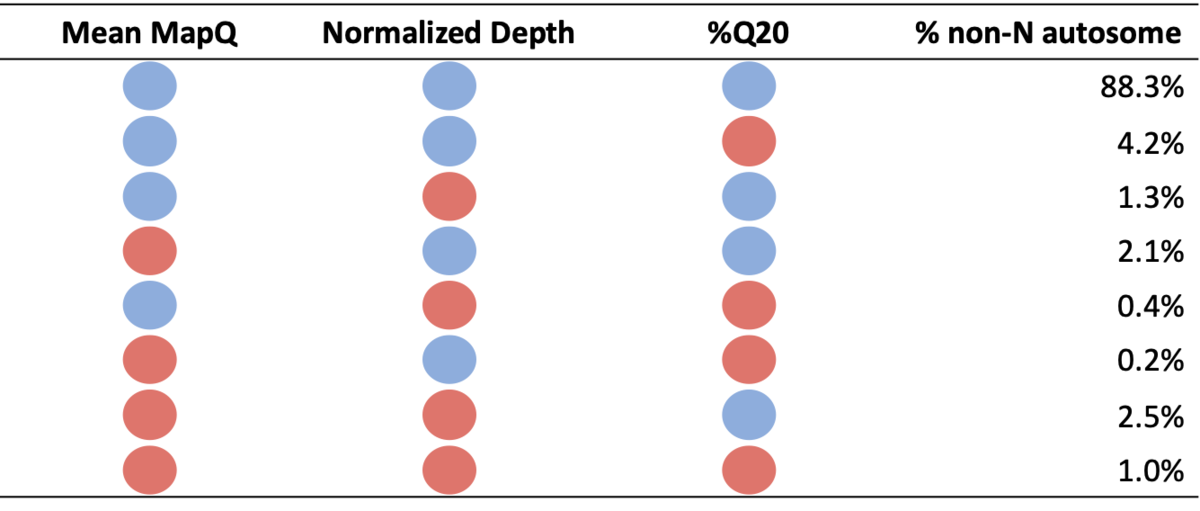
表1:N以外の常染色体が各系統的品質binに入る割合。青丸は特定のメトリクスに対する高い系統的品質を示し、赤丸は低い系統的品質を示します。
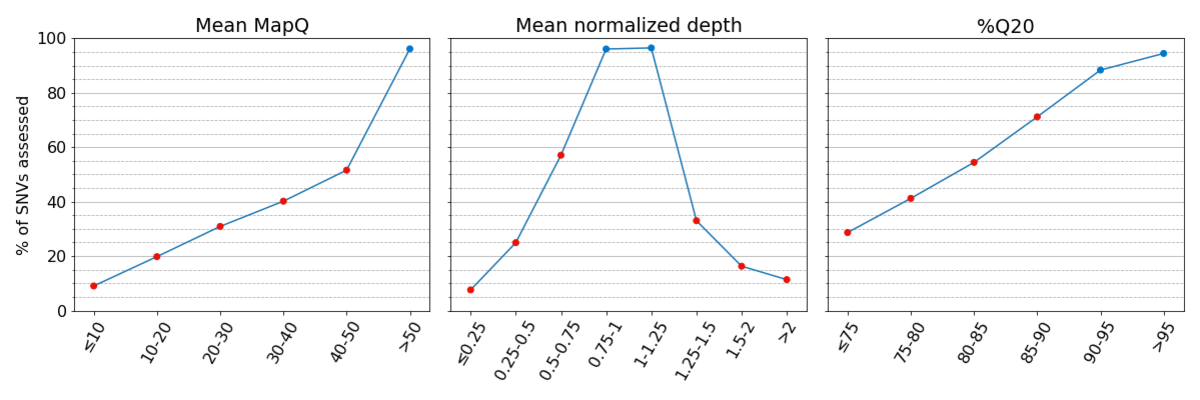
図2:真理値セットでは、系統的品質が低い領域では評価可能なSNVがほとんどありません。Platinum Genomes v2017.1の真理値セットについて、binあたりの評価済みSNVの割合を示します
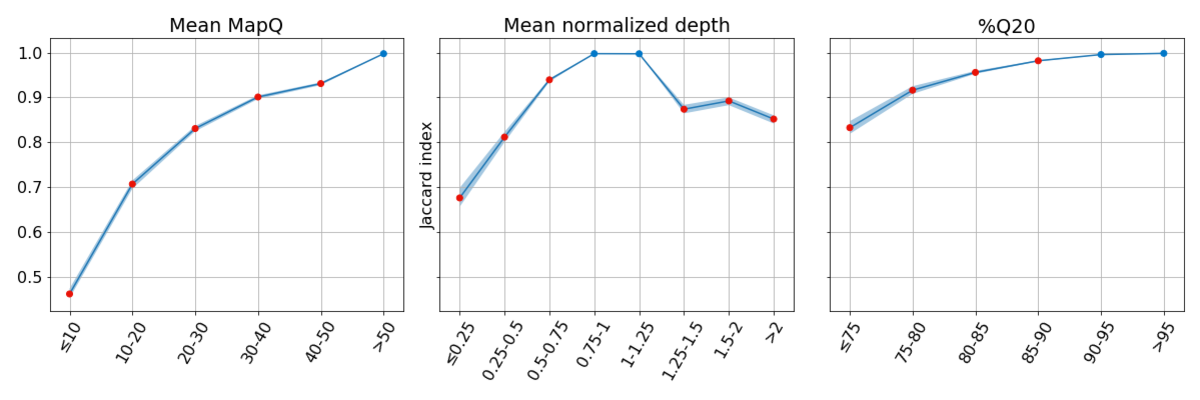
図3:系統的品質の低いゲノム領域では、一致率が下がります。各binでは、その他の2つのメトリクスが高い系統的品質を持つ座位で一致率が報告され、各メトリクスの系統的品質が低い影響が独立して浮き彫りになります。
Genome in a Bottleの困難領域との経験的系統的品質の比較
ゲノムのリファレンス機能はバリアントコーリング性能に悪影響を与える可能性がありますが、これらの領域のバリアントコーリング精度は明確に定義されていません。リファレンス機能のみに基づいてバリアントを除外すると、多くの高品質のバリアントコールが除外されることがあります。例えば、Genome in a Bottleは、低いマッピング可能性、セグメント重複、ロングタンデムリピート、極端なGC含有率の領域など、ゲノムの困難領域のいくつかのクラスを定義します(4)。これらの各クラスにおいて、この解析では、これらの困難領域の18~73%が高い系統的品質であるとラベル付けされています(表2)。当社の解析で系統的な品質が高いと判断された困難なクラスに分類される約61万8,000のSNVの一致率は98.8%です。さらに、GIABが困難領域と定義した範囲外にある約11万8,000のSNVで、当社の解析に基づく系統的品質が低い場合、一致率は79.9%です。これらの結果を総合すると、前述した困難領域が過度に広範であることが示され、経験的な系統的品質を使用することが生殖細胞系列バリアントコール性能のより良い予測因子となる可能性があることが示唆されます。
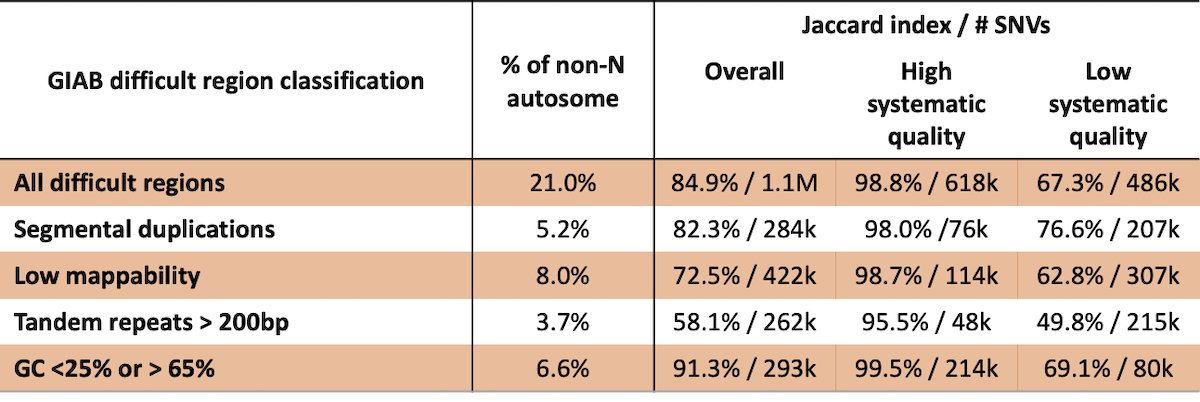
表2:GIABの困難領域は、バリアントコーリング性能の予測因子としては不十分です。
経験的な系統的品質と真理値セットの信頼度の高い領域の比較
真値値セットの信頼領域の特徴付けは、系統的品質のビニングから生成された領域と似ているが同一ではない、信頼度の高いバリアントコールに対応する領域を生成するはずです。さらに、これらの真理値セットが同定した信頼領域は、同じサンプルでも時間の経過とともに進化する可能性があります。これを調べるために、経験的に定義された信頼領域との重複に基づいて、異なる真理値セットの信頼領域の性能を比較しました(表3)。特に重要なのは、真理値セットの信頼領域内にあり、高い系統的品質を持つ座位が一貫して高い一致率(>99.9%)を示すことです。同様に、真理値セットの信頼領域に含まれていない系統的品質の低い座位では、一致率が非常に低くなります。
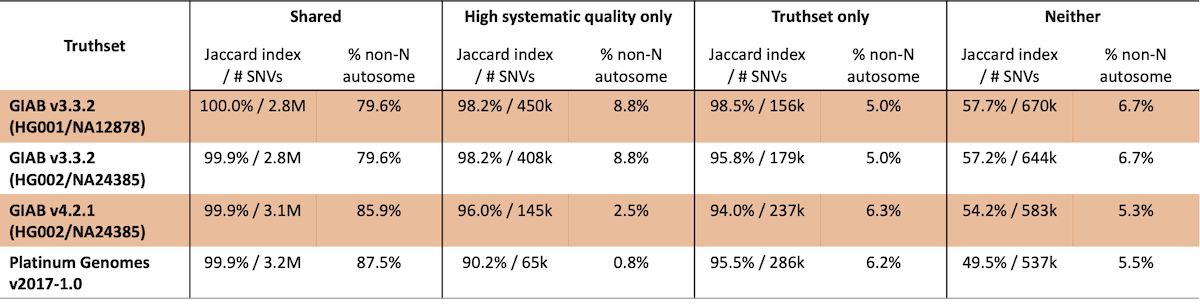
表3:系統的品質の高い領域と比較した、真理値セットのSNV一致率
この解析では、調査した真理値セットに存在しない65,000~450,000のSNVが同定され、一致率は約91%~>98%の範囲であったことから、これらの座位は一般的に高品質であることがわかります。これらの観察結果は、この解析がかなり未発達であるという事実と一致しています。当社の手法が発展するにつれて、系統的品質の高い座位における、さらなる一致率の向上が期待されます。
これらの観察結果は、さらに2つの結論につながります。第一に、ここで検討するbinの粗さを考慮すると、より高度な解析を通して、これらの真理値セットに固有のバリアントの一部が、最終的には高い系統的品質を持つものとして分類される可能性があります。ゲノムの残りの12%の一部のサブセットは、高い信頼度があるとみなされるために直交検証を必要とするバリアントコールを生成することができます。これはまさに真理値セットが提供するものです。家系の一貫性、直交プラットフォームとパイプライン、または手作業によるキュレーションのいずれを使用しても、この追加のエビデンスによりゲノムのより困難な領域で高品質のコールが可能になります。
ディスカッション
ゲノムデータの臨床的有用性の向上を成功に導くには、広範囲のゲノムワイドスケールだけでなく、高度にローカルなスケールにおいても、当社の性能を完全に理解することがきわめて重要です。大規模な集団にわたってゲノムワイドなデータを集約することで、あらゆる関心領域における当社製品の性能に関する詳細な情報の入手を始めることができます。重要な遺伝子への特定の影響が見られる場合、この情報はNGSで得られる情報を活用して臨床的に実行可能な結果を提供するための専門的なコーラーの開発に役立ちます。これは、イルミナのSMAおよびCYP2D6の専用コーラーで実証されています6,7。小規模バリアントコールの性能を詳しく見ていくことで、系統的エラーがアプリケーション固有の性能にどのように影響するか、ますます明確に把握することができます。これは、現時点で信頼性の高い臨床結果を提供できる場合を理解し、将来の臨床ゲノムを改善するために、次に重点を置く必要のある領域を明確にするための将来のアプリケーションを提案しています。
ここで示した結果は、バリアントコール性能を予測する機能に焦点を当てた、信頼領域の普遍的なセットを開発することが可能であることを示唆しています。当社は、系統的品質の高い領域において、ペアワイズレプリケートの一致率によって評価される性能が、SNVに対して非常に高いことを実証しました。重要な点は、系統的品質の高い領域が評価セットに対して別々のサンプルで定義されたため、これらの結果はシーケンスされたゲノム全体で一般化できる可能性が高いという点です。現在、この研究では、88%を超える常染色体においてSNVコールのレプリケーション率が約99.8%であることを確認しており、ゲノムのこの部分における直交検証の必要性がほとんど不要またはまったく不要であることを示しています。今後も、ゲノムのより大きな割合をカバーする信頼度の高い領域をより正確に同定するための作業を継続していきます。さらに、この作業ではSNVの一致率に焦点を当てましたが、より複雑なバリアントタイプや体細胞バリアントコーリングにも同じ手法を拡張できます。
学術用途向けの詳細情報またはDRAGEN試用版ライセンスについては、dragen-info@illumina.comまでお問い合わせください。
注釈
- Eberle MA, Fritzilas E, et al. A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res. 2017;27(1):157-164. doi:10.1101/gr.210500.116
- Wagner J, Olson ND, et al. Benchmarking challenging small variants with linked and long reads. bioRxiv. 2020;212712. doi:10.1101/2020.07.24.212712
- Zook JM, McDaniel J, et al. An open resource for accurately benchmarking small variant and reference calls. Nat Biotechnol. 2019;37:561-566. doi:10.1038/s41587-019-0074-6
- Krusche P, Trigg L, et al. Best practices for benchmarking germline small-variant calls in human genomes. Nat Biotechnol. 2019;37:555-560. doi:10.1038/s41587-019-0054-x
- Clarke L, Fairley S, et al. The international Genome sample resource (IGSR): A worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 2017;45(D1):D854-D859. doi:10.1093/nar/gkw829
- Chen X, Sanchis-Juan A, et al. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet Med. 2020;22:945-953. doi:10.1038/s41436-020-0754-0
- Chen X, Shen F, et al. Cyrius: accurate CYP2D6 genotyping using whole-genome sequencing data. Pharmacogenomics J. 2021;21:251-261. doi:10.1038/s41397-020-00205-5