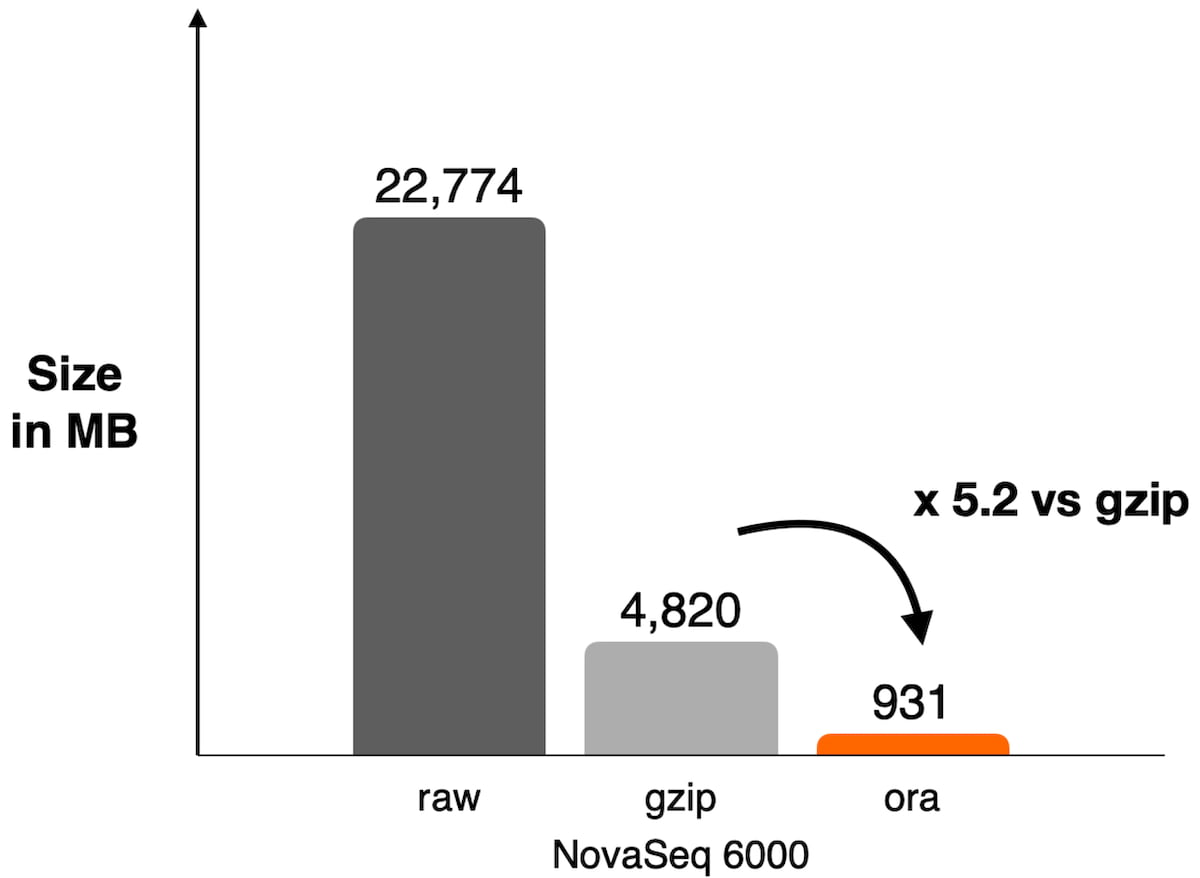
図1:NovaSeq 6000においてWESデータSRR8581228で使用したDRAGEN ORA圧縮で到達した圧縮率の例
ハイスループット次世代シーケンサー(NGS)で生成されるデータ量が飛躍的に増加するにつれ、FASTQ.ORA形式はストレージサーバーの維持に必要なストレージコストとエネルギーの両方を大幅に削減します。最大のインパクトを達成するには、この形式を可能な限り広く採用する必要があります。採用に有利になるように、この形式の仕様を公開しました(https://support-docs.illumina.com/SW/ORA_Format_Specification/Content/SW/ORA/ORAFormatSpecification.htm)。この記事では、NGSワークフローでの使用のために特別に設計された形式の特徴について説明します。
gzip圧縮とは異なり、DRAGEN ORAはゲノムデータに特化した圧縮テクノロジーです。高い圧縮率は、効率的な可逆エンコーディング方式によるものであり、本稿で詳しく解説します。
効率的なエンコーディングに加え、.ORA形式はユーザーが以下のような追加の利点を享受できるよう設計されています。
- ストリーミング。.ORAファイルをダウンロードする場合、リードの最初のブロックがダウンロードされるとすぐに解凍され、解析を開始できるため、ダウンロード全体が完了するまで待つ必要はありません。
- 圧縮ファイル連結。この形式では、複数の.ORAファイルを1つのファイルに書き込むことができます。この機能は、複数のFASTQ.ORAファイルの取り扱いを簡素化します。
- 並列圧縮/解凍。この形式の設計により、圧縮と解凍の両方のプロセスを加速するための、効率的なマルチスレッドによる実装が可能になります。
- メタデータの追加。この形式では、カスタムメタデータを追加できます。
- 暗号化。この形式は、適切なキーがない限りデータを読み取れないようにするため、オプションで暗号化レイヤーを追加することが可能です。
前文
この新しい圧縮形式の全体的な目的は、計算時間とコストを節約することです。すべての設計選択は、これを指針としています。
これにより、この形式は学術研究で開発された多くの圧縮プロトタイプとは異なります。学術的な研究では、圧縮率の限界を追求することが多く、計算コストを度外視して極限まで圧縮率を高めたり、新しい圧縮手法の理論的可能性を探求したりすることが一般的です。例えば、Assembltrie1や lfqc2 は、高い圧縮率を実現することを目的としていますが、計算コストも高くなります。
1 ORA圧縮形式の目的
ORA圧縮形式には、完全可逆とNGSワークフローへの低コストの統合という2つの必須要件があります。
1.1 完全可逆圧縮
圧縮形式には、非可逆圧縮と可逆圧縮があります。例えば、オーディオ、画像、または動画の場合、より大きな圧縮を可能にするためには、元のデータを変更することが許容されます。データ変更は必ずしもユーザーが認識できるわけではないため、これは理にかなっています。ゲノムデータの場合、圧縮データで実施される解析は、非圧縮データと同じ結果を得る必要があります。二次解析を開始する前に、ユーザーはこの要件が満たされていることを確信できなければなりません。これを実現する最善の方法は、完全な可逆形式を採用することです。つまり、圧縮・解凍のサイクルを経てもデータがバイト単位で完全に保持される必要があり、MD5などのチェックサムを用いることで、その可逆性を容易に検証できるようになります。
SPRING3やORCOM4などの既存のFASTQ圧縮形式は、圧縮を促進する効果的な方法であるため、リードの順序を変更します。しかし、これらの形式の中には、FASTQの初期リード順序が意味を持たないという前提のもとで、可逆(ロスレス)であると主張しているものもあります。たとえその前提が正しいとする議論があったとしても、チェックサム検証が利用できなくなるため、DNA配列が保持されていることを保証できず、DNA配列分野において情報が失われていないことを証明するのが困難になります。さらに、チェックサムの保持は多くの場合、品質および規制上の要件です。
これらの理由から、当社はリードの順序を変更する方法を選択せず、チェックサムの保持を必須としました。
1.2 NGSワークフローへの低コストの統合
コストの節約が主な要件であるため、圧縮率が非常に高いが、大量のメモリやコンピューティングリソースを必要とする圧縮形式は使えません。当社の圧縮形式は、不当な追加コストを生じさせず、既存の解析ワークフローで可能な限り混乱を起こさないようにする必要があります。
これは以下を意味します。
- メモリ使用量が少ない(圧縮/解凍がその他のプロセスと並行して発生する場合、圧縮の目的のみにより大きなシステムが必要とならない)
- ワークフロー全体を著しく減速させないほどのスピード
- 一時的なディスクファイルなしでその場で圧縮/解凍可能
- 大規模ファイルへの拡張が可能
1.3 高い圧縮率
最初の2つの要件(完全に可逆的で低コストの統合)が満たされている限り、もちろん、圧縮率をできるだけ高くしたいと考えています。
指摘事項
この分野の学術研究では、FASTQファイルの圧縮はリファレンスフリーの手法(リード間の類似性を探索する)またはリファレンスベースの手法(リードとリファレンスゲノム間の類似性を探索する)で実施できることが示されました。
リファレンスフリーの手法は、特にリードの再配置手法と組み合わせることで高い圧縮率を達成できる一方で、実行時間の増加やメモリ使用量の増大といった課題を伴う傾向があります3、4。
FQZip5やLW-FQZip6など、リファレンスベースの手法は原則としてより容易です。ゲノムにリードをアライメントし、位置と差異のリストをエンコードします。また、高い圧縮率を達成するためにリードの順序を変更する必要はありません。これらは、良質なリファレンスを利用できる場合にのみ可能です。作成および保管されるデータの大部分はヒトのデータであり、今後もヒトのデータとなるため、非常に良好なリファレンスが利用できるリファレンスベースの手法を選択することは理にかなっています。
当社は、要件を考慮して、リファレンスベースの圧縮法を選択しました。これは最も実用的な選択のようです。ヒトのリファレンスゲノムを用いて生成されたゲノムデータの大部分をカバーしながら、高速な実行と高い圧縮率を可能にします。
2 FASTQ.ORA圧縮形式の説明
多くの設計選択は、処理速度と圧縮率のバランスを最適化するために行われました。この形式は、イルミナのシーケンスデータの特性である、低いシーケンスエラー率、Indelエラーなし、ショートリードを考慮して最適化されています。
.ORA圧縮形式は、すべてのイルミナプラットフォームで生成されるFASTQデータで機能しますが、Novaseq 6000やNextSeq 1000/2000などの最近のイルミナプラットフォームで最も効率的です。
2.1 形式レイアウト
ORAファイルはORAブロックのリストで構成されています(図2)。各ブロックには大量のリードが含まれており、解凍に必要なすべての情報を含む自己完結型のユニットです。したがって、全体的なファイルヘッダーはありません。各ブロックには独自のヘッダーセクションがあります。このスキームにより、圧縮ファイル連結、並列圧縮/解凍、およびストリーミング機能(ブロックが転送されるとすぐに解凍を開始)が可能となります。
各ブロックには、元のファイルと同じ順序で大量のリードが格納されます。ブロックはヘッダーから始まり、データセクションのリストが続きます。ヘッダーにはメタデータと各データセクションのサイズが格納されます。DNA配列、クオリティスコア、リード名には別々のデータセクションがあります。
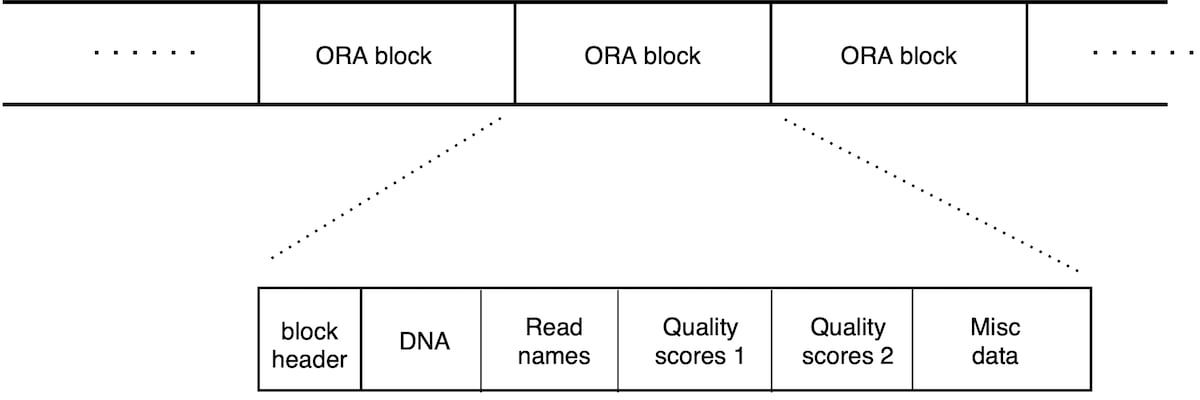
図2:.ORAファイルのレイアウト。それぞれ50,000リードを含むブロックのリスト。各ブロックは個別に解凍できます。各ブロックはヘッダーから始まり、DNA配列、リード名、およびクオリティスコア(以下に説明するように2つのセクションに分割)の異なるデータセクションが含まれます。
図2のORAファイルのレイアウトは、それぞれ50,000リードを含む一連のブロックを示しています。各ブロックは個別に解凍できます。各ブロックはヘッダーから始まり、DNA配列、リード名、およびクオリティスコア(以下に説明するように2つのセクションに分割)の異なるデータセクションが含まれます。
2.2 ORAブロック
2.2.1 DNA配列のエンコーディング
リファレンスベースのコンプレッサーがあるため、リードはまずリファレンスゲノムにマッピングされ、次にゲノム内の位置と差異のリストとしてエンコードされます。マッピングアルゴリズムは形式仕様の一部ではなく、実装に依存します。注意すべき点は、リードアライナーの選択は、圧縮速度と圧縮率において重要な役割を果たしていることです。ブロックヘッダーのビットフラグは、ブロック内のリードがすべて同じサイズであるかどうかを示します。そうでない場合、サイズのリストが別のデータセクションに保存されます。
配列はカスタムバイナリ形式でエンコードされ、エントロピーコーダーであるzstdライブラリーでさらに圧縮されます7。
リードは、リファレンスゲノム上の位置(32ビットの絶対位置または可能な場合は16ビットの相対位置を使用)と、必要に応じてミスマッチのリストでエンコードされます。各ミスマッチは1バイトでエンコードされ、ミスマッチの6ビット相対位置と2ビットの代替ヌクレオチドが加わります。相対位置を大きくする必要がある場合は、間に「偽の」ミスマッチを追加します。クリップされた部分は許容されており、配列の内容に応じて、1塩基あたり2ビットまたは4ビットでエンコードされます。マップされていないリードは未処理のまま、ヌクレオチドあたり2または4ビットでエンコードされます。
シンプルさと速度のために、Indelシーケンスエラーは使用しません。最適なアライメントにIndelが必要な場合は、リードはより多くのミスマッチまたはクリップされた部分を使用してエンコードされます。イルミナのシーケンスデータでは、Indelシーケンスエラーはまれであるため(0.03%未満)、圧縮率に有意な影響はありません。最も重要なのは、Indelを省略することで、非常に高速なマッパーを使用できることです。
ORA形式では、リードに4つの異なるエンコーディングを使用します。位置のみを必要とする完全にマッピングされたリード、位置+ミスマッチリストを必要とするグローバル配列、位置+ミスマッチリスト+クリップシーケンスを必要とするローカル配列、およびマップされていないリードです。一部のフラグは、各リードに対して、使用されたエンコーディングの種類、位置の形式(絶対または相対)、ミスマッチの数、およびクリップされた部分の長さを示します。
例として、一部のエンコーディングのビットサイズのリストを以下に示します。
- 相対位置での完璧な配列:4フラグビット + 16ポジションビット = 合計20ビット
- 絶対位置での完璧な配列:4フラグビット + 32ポジションビット = 合計36ビット
- グローバル配列、2ミスマッチ、絶対位置:12フラグビット + 32ポジションビット + 2x8のミスマッチビット = 合計60ビット
- ローカル配列、2ミスマッチ、クリップされた部分10ヌクレオチド:合計92ビット
- マップされていない150ヌクレオチドのリード:300ビット
このバイナリ形式でリードをエンコードした後、zstdライブラリー7を使用してさらに圧縮します。
圧縮後(バイナリエンコーディング + zstd)、各ヌクレオチドは平均0.3~0.5ビットでコードされます(生FASTQではヌクレオチドあたり8ビット、gzip圧縮FASTQでは約2ビット/ヌクレオチド)。
2.2.2 クオリティスコアのエンコーディング
最近のシーケンサーからのデータの場合、クオリティスコアは最大4つの異なる値しか必要とせず、値0は未確定(N)塩基のクオリティスコアに使用されます。最初のステップとして、'N'を含まないリード(3種類のクオリティ値のみを持つ)と、それ以外のリードを区別します。その結果、クオリティスコアは2つのグループに分類され、これらは図2に示されている2つのクオリティスコアセクションに対応します。
リードの大部分を表す3つの異なるクオリティ値を持つグループでは、まずクオリティ値ごとに1.6ビット(3種類のためlog2(3) ≈ 1.58)でクオリティ値を再エンコードします、次に、範囲エンコーダを使用し、直前の30ウィンドウのコンテキスト内における最高値のクオリティスコアの数を表す4ビットの値コンテキストモデルを適用します。
シーケンサーのクオリティ値のビニングスキームは、図3に示すように、クオリティフィールドが圧縮ファイル内で消費する相対的なスペースに影響を与えます。また、図4に示すように、圧縮率にも影響を及ぼします。
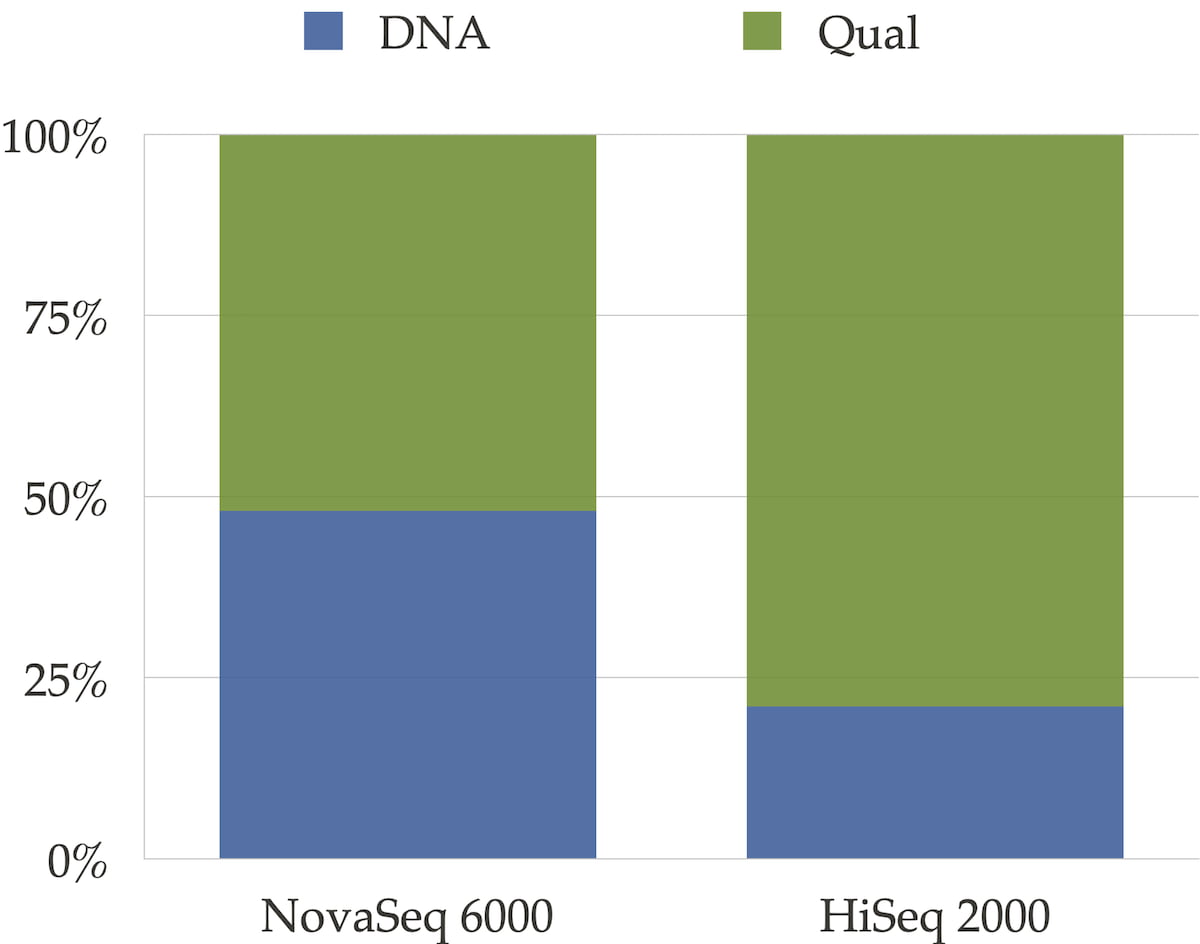
図3:NovaSeq 6000およびHiSeqTM 2000のデータタイプにおけるDNAデータとクオリティデータの分布 HiseqTM 2000装置では、クオリティスコアが40の異なる値にエンコードされており、クオリティデータがファイルサイズの大部分を占めます。NovaSeq 6000の場合、クオリティデータとDNAはほぼ同じ容量となります。
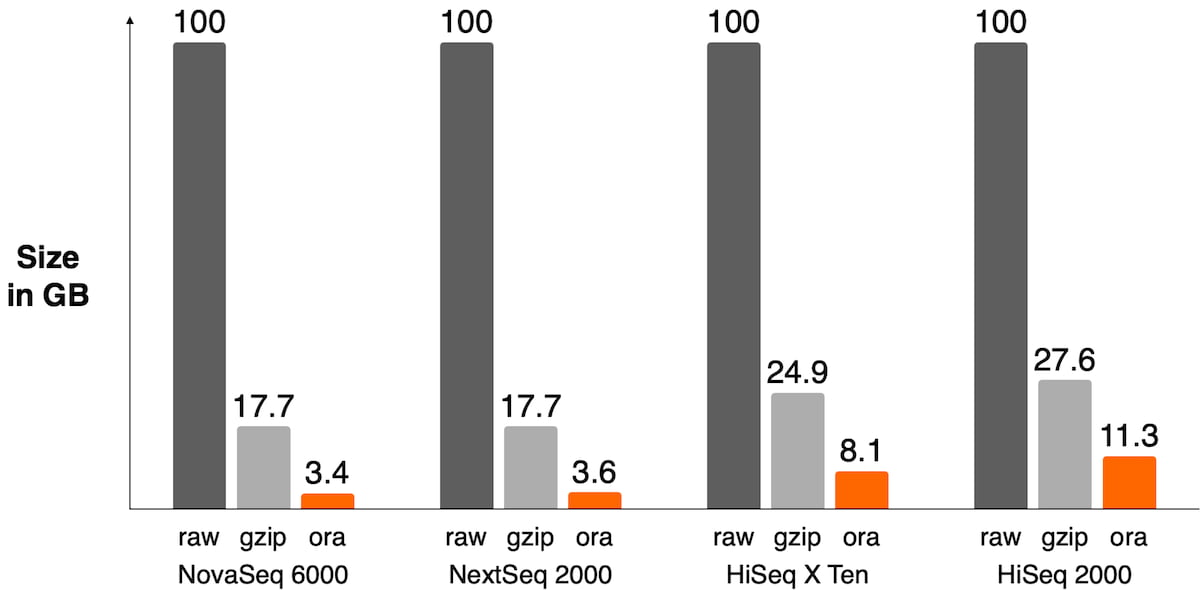
図4:さまざまなシーケンサー上の生FASTQおよびgzip圧縮ファイルに対するFASTQ.ORAファイルのサイズ。DRAGEN ORAは、NovaSeq 6000やNextSeq 1000/2000など、クオリティスコアが2ビットでエンコードされている最近のシーケンサーで最高の圧縮率を実現します(gzip圧縮ファイルに対して4~5倍の圧縮率)。古いシーケンサーでは、クオリティスコアの可逆圧縮がファイル内の多くのスペースを占め、全体的な圧縮率を大幅に低下させます。
2.2.3 リード名のエンコーディング
リード名はトークン化され、数字と非数字のフィールドを分離します。その後、各トークンはブロック内のすべてのリードに対して「列単位」で圧縮されます。数字フィールドは、前のリードからのオフセットとしてエンコードされ、zstdライブラリーでは数字以外としてエンコードされます。
いくつかの既存のFASTQ圧縮形式は、すでにこのスキームのバリエーションのひとつを使用しており、シンプルかつ効率的であることが示されています(例:Fqzcomp8)。
2.2.4 整合性チェック
整合性チェックは圧縮形式の重要な部分です。ORA形式では、圧縮ファイルの各ブロック内に2つのチェックサムが保存されます。
- 圧縮データのチェックサム:これにより、ファイルが解凍される前にファイルの破損を確認することができます(圧縮されたファイルは圧縮後に変更されたか否か)。
- 元の非圧縮データのチェックサム:解凍時に可逆的に解凍することを検証できます。
整合性チェックは、ユーザーが手動で実行できるようになっています(例:gzip --testと同様)。当社の実装では、ファイルが圧縮されるたびに常に完全な整合性チェックが自動的に実行されます(新しい圧縮ブロックが生成されると、すぐに解凍され、インプットと比較されます)。
2.2.5 リファレンスゲノム
現在DRAGEN ORAで使用されている初期設定のヒトリファレンスゲノムはhg38です。
このリファレンスは圧縮ツール固有のものであり、解析に使用できるリファレンスツールとは完全に独立しています。二次解析のマッピングには、圧縮とは完全に異なるリファレンスゲノムのバージョンを使用する可能性があります。
さらに、.ORA形式では、必要に応じて他のリファレンスゲノムを使用できます(ただし、解凍時に圧縮時と同じリファレンスが必要になります)。
3 結果的な性能/設計の特徴
DRAGEN ORA圧縮ツールは、上記の厳しい要件を満たすように設計されています。この設計は高性能につながり、以下に説明するさまざまな便利な機能を可能にします。
3.1 bzip2 / SPRINGとの比較
表1では、DRAGEN ORAをbzip2およびSPRING3と比較します。ここでは包括的な比較を行うつもりはなく、より高い圧縮率を達成するgzipの一般的な代替手段であるbzip2と、公開されている最新のFASTQ圧縮形式の1つであるSPRINGを選択しました。SPRINGは初期設定オプションで使用しました。
試験は16スレッドで実施されます。入力としてFASTQ.GZファイルから圧縮し、FASTQに解凍します。
表1:さまざまなサイズのファイルでbzip2、gzip、SPRINGと比較したDRAGEN ORAの性能
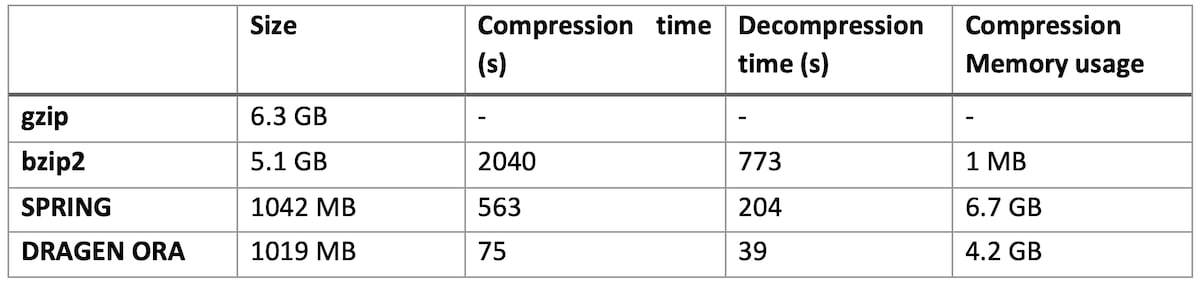
ファイルSRR9613620(ヒトエクソームシーケンスデータ、FASTQ.GZ 6.3 GB)
ファイルSRR9273189(ヒトエクソームシーケンスデータ、FASTQ.GZ 14.4 GB)
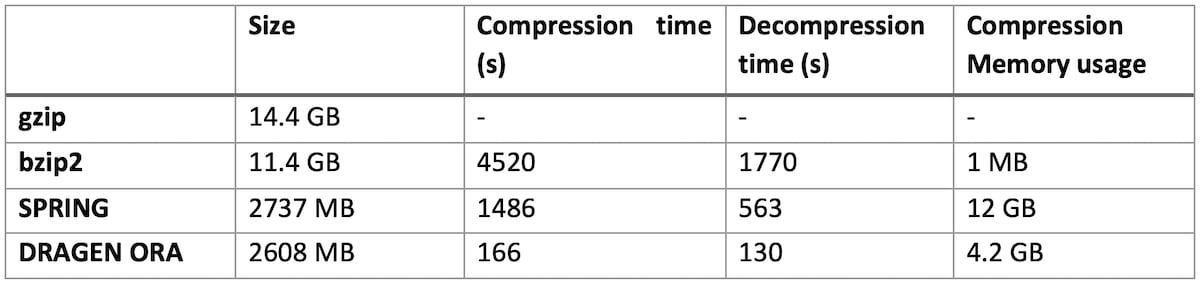
DRAGEN ORAは一貫して高速で(上記の試験ではSPRINGより7倍以上速い)、SPRINGよりも小さなファイルサイズです。
特に、DRAGEN ORAのメモリ使用量はファイルサイズに依存しません。一方、SPRING3などのその他の圧縮方法では、入力ファイルのサイズに応じてメモリ要件が増大するため、大きなファイルでの使用には負担があります。
3.2 pigz/gzipと比較した圧縮/解凍速度
表2に示す実験では、DRAGEN ORAの圧縮/解凍速度を、7 GBのFASTQ.GZファイルで16スレッドで実行し、gzipの並列バージョンであるpigzと比較します。この試験では、DRAGEN ORAの圧縮はFASTQ.GZからFASTQ.ORA、解凍はFASTQ.ORAからFASTQです。
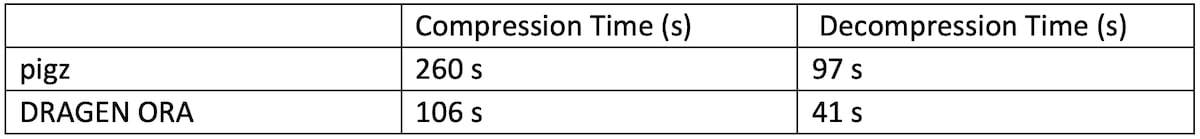
表2:pigzと比較したDRAGEN ORA速度性能
DRAGEN ORAは、圧縮と解凍の両方において、pigzよりも高速です。
DRAGEN ORAの圧縮(FASTQ.GZ -> FASTQ.ORA)では、入力ファイルを圧縮する前に解凍する必要があるため、解凍に要する時間がDRAGEN ORA圧縮処理時間の下限となります。
DRAGEN ORAの圧縮時間は106秒であり、16スレッドで実行した場合の下限値(97秒)に非常に近いことが確認できます。
3.3 形式仕様によって可能になる機能:ストリーミング、メタデータ、暗号化
.ORA形式は、ストリーミング用途に適した設計となっています。.ORAファイルをダウンロードする場合、最初のブロックが受信され次第、解凍処理を開始できます。逆に、ローカルファイルを圧縮する場合、最初のブロックが圧縮され次第、すぐに圧縮ファイルのアップロードを開始できます。DRAGEN v 3.9では、AWS S3またはAzure Blob StorageからORAファイルをストリーミングできます。
さらに、.ORA形式はAESを使用した暗号化をサポートできるように設計されており、ORA実装ではORAアーカイブにカスタムメタデータを書き込むことができます(https://support-docs.illumina.com/SW/ORA_Format_Specification/Content/SW/ORA/ORAFormatSpecification.htm)。注:現在、これらの機能は、.ORA形式への圧縮の現在の実装(本稿の発行時点ではDRAGEN 3.9)では実装されていません。
ここでは、.ORA形式がハイスループットのゲノムデータの管理要件に十分対応していることを示しています。ゲノミクス分野は、これを使用することによって大きな恩恵を受ける可能性があります。手法を説明し、仕様を共有することで、その採用を促進したいと考えています。
謝辞 Michael Ruehle氏とRami Mehio氏のご支援と本稿の査読に感謝いたします。
学術用途向けの詳細情報またはDRAGEN試用版ライセンスについては、dragen-info@illumina.comまでお問い合わせください。
注釈
- Ginart, A. A., Hui, J., Zhu, K., Numanagić, I., Courtade, T. A., Sahinalp, S. C., & David, N. T. Optimal compressed representation of high throughput sequence data via light assembly. Nat Commun 9, 566 (2018)
- Nicolae, M., Pathak, S., & Rajasekaran, S. LFQC: a lossless compression algorithm for FASTQ files. Bioinformatics 31, 20, 3276-3281 (2015)
- Chandak, S., Tatwawadi, K., Ochoa, I., Hernaez, M., & Weissman, T. SPRING: a next-generation compressor for FASTQ data. Bioinformatics, 35, 15, 2674-2676 (2019)
- Grabowski, S., Deorowicz, S., & Roguski, Ł. Disk-based compression of data from genome sequencing. Bioinformatics 31, 9 (2015), 1389-1395
- Zhang, Y., Li, L., Xiao, J., Yang, Y., & Zhu, Z. FQZip: lossless reference-based compression of next generation sequencing data in FASTQ format. Proceedings of the 18th Asia Pacific Symposium on Intelligent and Evolutionary Systems (2015) Volume 2 (pp. 127-135). Springer, Cham. DOI: 10.1007/978-3-319-13356-0_11
- Huang, Z. A., Wen, Z., Deng, Q., Chu, Y., Sun, Y., & Zhu, Z. LW-FQZip 2: a parallelized reference-based compression of FASTQ files. BMC bioinformatics, 18, 1, 1-8 (2017)
- https://tools.ietf.org/id/draft-kucherawy-dispatch-zstd-00.html
- Bonfield, J. K., & Mahoney, M. V. Compression of FASTQ and SAM format sequencing data. PloS one, 8, 3, e59190 (2013)