はじめに
脊髄性筋萎縮症(SMA)は、アルファ運動ニューロンの喪失を特徴とする常染色体劣性神経筋疾患であり、出生時または出生直後に見られる重度の筋力低下と萎縮を引き起こします1。SMAは嚢胞性線維症に続き、乳児死亡の主な遺伝的原因です。SMAの発生率は出生6,000~10,000人に1人で、キャリア頻度は民族により異なり1:40~1:80です2–4。SMA症状の緩和に対してFDAの承認を受けているNusinersen5とZolgensma6という2種類の早期治療が利用できるため、SMAの早期発見は長期的な生活の質にとって極めて重要です。
疾患原因遺伝子であるSMN1は、そのパラログ遺伝子SMN2とともに、5番染色体の約2Mbの領域に存在し、多数の複雑なセグメント重複と逆セグメント重複を伴います。SMN2は、ヒト系統に特有の先祖遺伝子の重複によって作製されました7。SMN1とSMN2の周囲のゲノム領域は、不均等なクロスオーバーと遺伝子変換を受け、SMN1とSMN2のコピー数(CN)が変動します。重要な点として、SMN2はSMN1と>99.9%の配列一致率を持ち、その塩基の違いの一つであるエクソン7の c.840C>Tは、重要な機能的影響をもたらします。c.840Tは、スプライシングエンハンサーを中断することでエクソン7のスキップを促進し、SMN2由来の転写産物の大部分が不安定となり、完全には機能しません8。SMA症例の約95%は、SMN1の欠失またはSMN2への遺伝子変換(c.840T)によって引き起こされる機能性c.840Cヌクレオチドの両アリル欠損に起因しています9。SMAの残りの5%の症例では、SMN1にその他の病原性バリアントも見られます10。SMN2は少量の機能タンパク質を産生することができ、個人のSMN2コピー数は疾患の重症度と逆相関します11。
発生率と疾患重症度が高いため、米国医学遺伝学会では集団全体のSMAスクリーニングが推奨されています。SMAのスクリーニングの鍵は、1)SMA診断およびキャリア検査用のSMN1のコピー数の判定、2)臨床分類および予後用のSMN2のコピー数の判定です。従来、SMA検査とキャリア検査は、定量的PCR(qPCR)、マルチプレックスライゲーション依存性プローブ増幅(MLPA)、デジタルPCRなどのポリメラーゼ連鎖反応(PCR)ベースのアッセイで行われます。これらの方法では、主にSMN1とSMN2の間で異なるc.840C>T部位に基づいてSMN1のコピー数を判断します。WGSによるSMA検査を可能にすることは、プレシジョンメディシンのイニシアチブに大きく貢献しますが、SMN1とSMN2の間のほぼ完全なシーケンス同一性、およびハイブリッド遺伝子につながる2つの遺伝子間の頻繁な遺伝子変換などの課題に直面しています。これらの課題には、この分野の困難を克服するために特別に設計されたインフォマティクス手法が必要です。
この問題を解決するために、WGSデータに基づいてSMN1とSMN2の両方のCNを検出する新しい手法であるSMNCopyNumberCallerを開発しました12。この手法は、1000 Genomes Project(1kGP)の集団データを用いて開発されました13。この手法の発表で当社は、12,747のゲノムでSMN1とSMN2の特性評価を行い、1568件のサンプルでSMN1の増減があり、6,615件のサンプルでSMN2の増減があることがわかり、汎民族的なキャリア頻度は2%と計算され、これは過去の研究と一致しました。さらに、SMN1 CNコールの99.8%とSMN2 CNコールの99.7%が直交法と一致し、SMAでは100%、保因者では97.8%のコール率となり、精度はSMAとキャリアの両方で100%でした。このコーラーが、新生児ケアにおける包括的な検査として、またWGSシーケンスプロジェクトにおける正確なキャリアスクリーニングツールとして、SMA検査の提供を可能にします。
ここでは、多民族のサンプルを用いて、アフリカの集団において遺伝的変動性が高いことを同定でき、SMN1とSMN2を確実に区別できない可変部位を除外し、すべての集団で精度を最適化できた方法について説明します。これは、新しいインフォマティクス手法を開発する際に、民族的に多様な集団を使用することの重要性を強調しています。さらに、これらの遺伝子で行われたコピー数コールを裏付けるエビデンスをユーザーが評価できる静止画像を生成する視覚化ツールについて説明し、提示します。この情報は、WGSベースのSMAコールの実施を希望する臨床検査室にとって不可欠なツールとなります。
SMNCopyNumberCaller
SMN1とSMN2の全体的な高い配列類似性という課題に対応するため、SMNCopyNumberCallerはまず、両遺伝子のいずれかにアラインされたリードのシーケンスカバレッジを解析し、SMNと総称されるSMN1とSMN2の合計コピー数を算出します。エクソン7~8が欠失しているSMN2の短縮型(SMN2∆7-8と呼ばれる)があるため、完全型のSMNと短縮型のSMNのコピー数を計算する時に、遺伝子の領域をエクソン1~6を含む22.2kb領域とエクソン7~8を含む6.3kb領域の2つの領域に分割して計算します。エクソン7~8領域から計算されたCNは、完全型のSMN遺伝子の数を提供します。SMN2∆7-8のサンプルは、エクソン7~8領域のCNと比較してエクソン1~6領域のCNが高く、この差はSMN2∆7-8のCNを表しています(図1)。
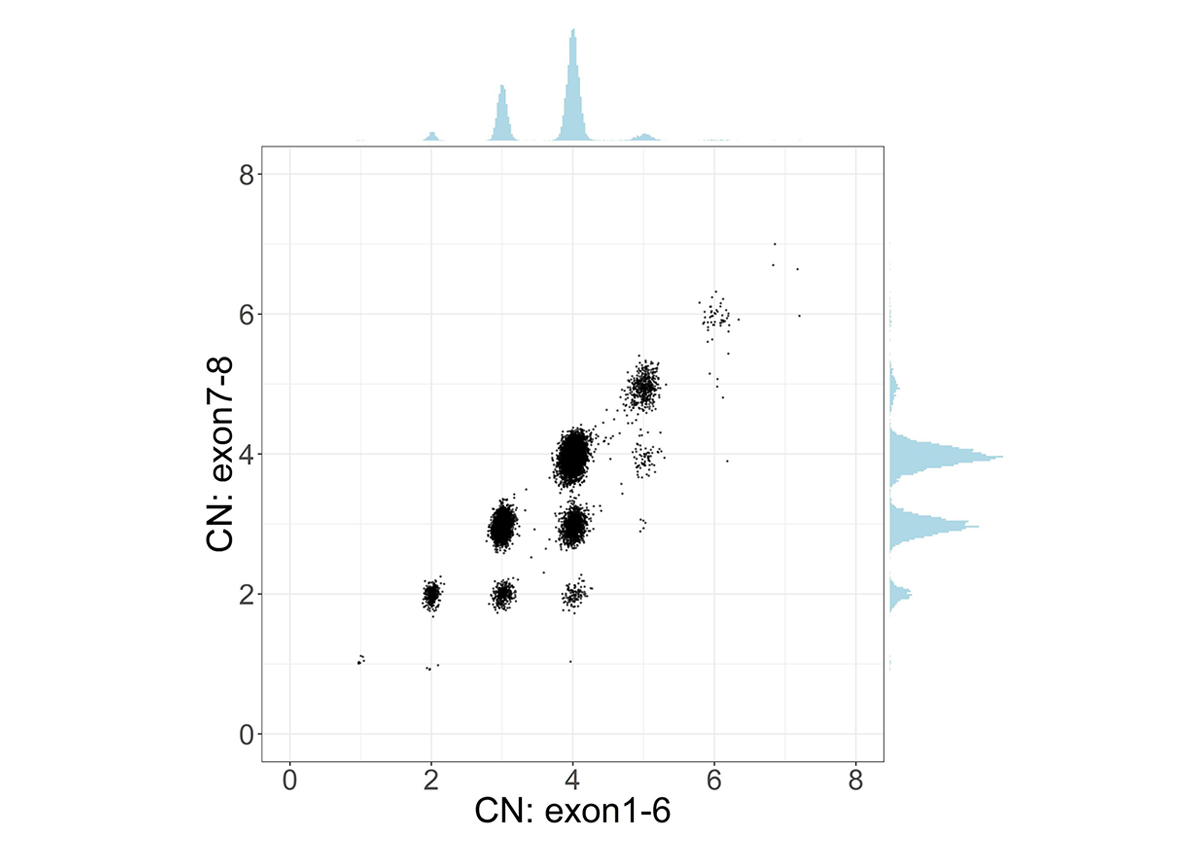
集団における全SMN(SMN1 + SMN2)コピー数(x軸、エクソン1~6のリード深度によってコールされる)および完全型SMNコピー数(y軸、エクソン7~8のリード深度によってコールされる)の散布図とヒストグラム。対角線下のクラスターは、エクソン7~8が欠失した遺伝子(つまりSMN2∆7-8)の存在を示します。
合計コピー数を算出した後、SMN1とSMN2の塩基配列の違いに基づき、それぞれの支持リード数を解析することで、 SMN1とSMN2を識別します。各部位におけるSMN1(SMN2)の個々のCNは、合計のSMN CNとSMN1+SMN2を支持するすべてのリードの中でのSMN1(SMN2)支持リードの割合を考慮して計算します。このコーラーの開発過程において、1kGPサンプルを用いて、リファレンスゲノムから抽出したSMN1とSMN2の16箇所の塩基差異部位におけるコピー数(CN)をコールし、それぞれの位置でのCNコールがc.840C>T スプライスバリアント部位でのCNコールと一致するかを検証しました。アフリカ人集団と非アフリカ人集団におけるコールの一致率には顕著な差が見られました(図2)。アフリカ人のサンプル以外では、スプライスバリアント部位とのCN一致率が高い(>85%)部位が13箇所ありました。逆に、アフリカ人のサンプルでは、スプライスバリアント部位とのCNの一致率が高い部位は7箇所のみで、非アフリカ人集団よりもすべての部位で一致率の値が低くなりました。これは、アフリカ人集団においてこれらの多くの部位で見られる遺伝子内変異と一致しています。この解析は、ゲノムの臨床的に意義のある難解領域を解決するための新しいインフォマティクス手法を開発する際、民族的に多様な集団を使用することの重要性を強調しています。
アフリカ人と非アフリカ人の両集団において、スプライスバリアント部位と高度に一致した7つの部位と、スプライスバリアント部位を含む8つのSMN1/2塩基の違いを選択しました。これらの部位のみを選択することで、このコーラーは民族性に関係なく、一貫して性能を示すはずです。SMNCopyNumberCallerは、8つの選択された部位でのCNコールの一致に基づいてSMN1およびSMN2 CNコールを行います。
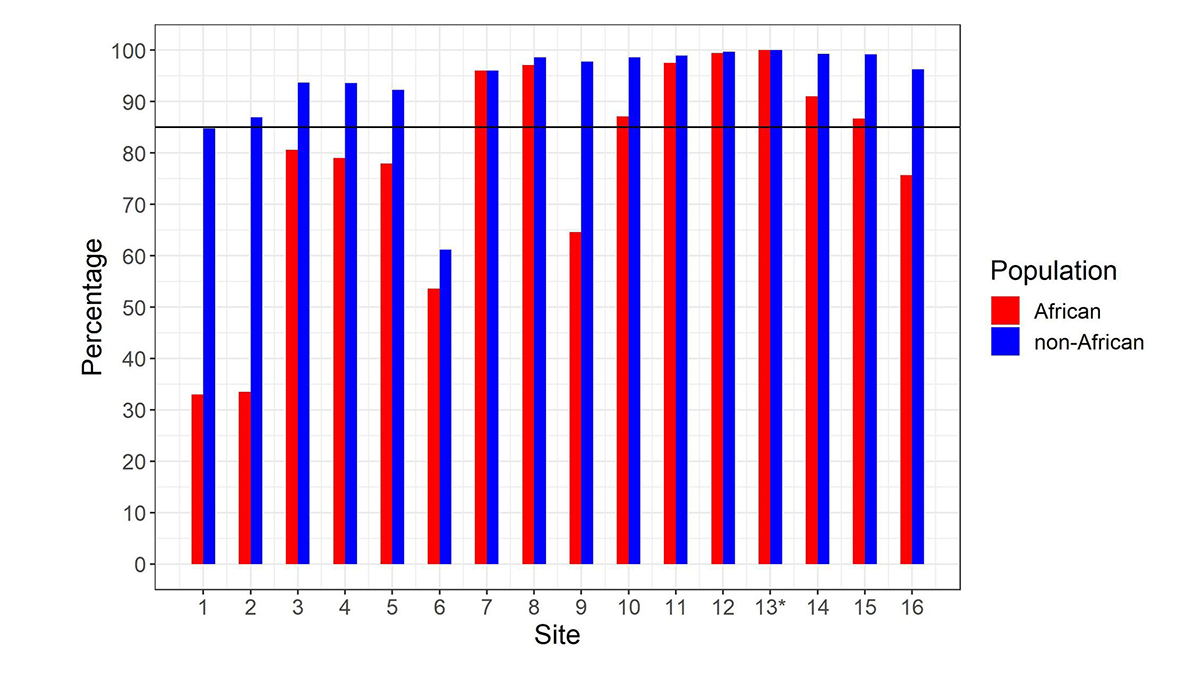
アフリカ人および非アフリカ人集団における16のSMN1~SMN2塩基差異部位におけるc.840C>Tとのコピー数(CN)コールが一致したサンプルの割合。部位13*はc.840C>Tスプライスバリアント部位です。黒の横線は85%の一致率を示します。SMNCopyNumberCallerは、アフリカ人と非アフリカ人の集団(7、8、10、11、12、13、14、15)の両方で85%を超える一致のある8つの部位のみを使用します。これらの8つの部位の座標(hg38、chr5)は、70950493、70950966、70951392、70951463、70951897、70951946、70952094、70952209です。
コーラーの結果の可視化
臨床現場でのバリアントコールの重要なコンポーネントは、臨床レポートに署名する際に裏付けとなるエビデンスを確認する必要があることです。SMNCopyNumberCallerのリリース以降、データを表現し、コールの品質管理(QC)を行うための静的画像を生成する視覚化ツールを開発しました(図3)。全SMN(エクソン1~6、図3A)および完全型SMN(エクソン7~8、図3B)の合計CN(SMN1+SMN2)は、集団分布に対してプロットされます。全SMN CNと完全型SMN CNの差は、SMN2∆7-8のCNを表します。SMN1とSMN2の個々のCNは、SMN1とSMN2の8塩基差異(図3Cの#7-8と#10-15)における完全型CNの合計と支持リード数に基づいて計算されます。図3Dは、分化部位でのリード数のみに基づくSMN1およびSMN2のコピー数の概算値を示しています。
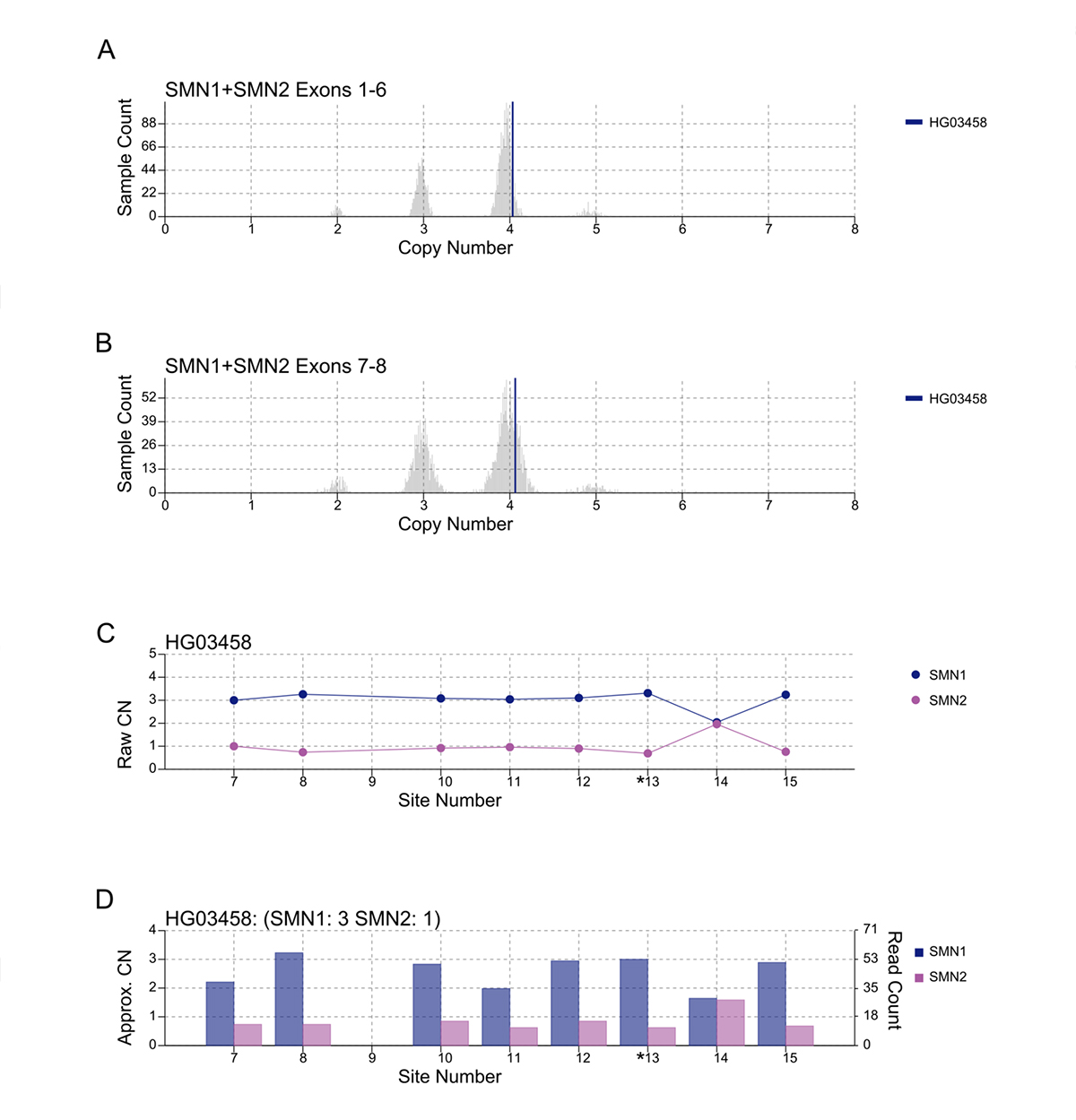
A/B. 全SMNコピー数(A)および完全型SMNコピー数(B)の1kGP集団サンプルに対する生データの深度値(垂直線)。C. SMN1およびSMN2の8箇所の部位(#7-8、#10-15)における生データのコピー数(CN)値。これらの値を基にコンセンサスを決定します。各部位におけるSMN1(SMN2)の生データのコピー数(CN)は、完全型SMNのCNにSMN1(SMN2)の支持リード数の割合(SMN1 + SMN2の支持リード数に対する比率)を掛けることで算出されます。*13番はスプライスバリアント部位です。D. SMN1およびSMN2の生データのリード数は右のy軸に表示されます。左のy軸はCNの概算値を示しており、リード数をサンプルのハプロイド深度の中央値で割った値で推定されます。
性能検証
この手法の精度を実証するために、デジタルPCRとMLPAを使用したCNコールをWGSベースのコールと比較し、SMN1では99.8%、SMN2では99.7%の一致率を示し、SMAでは100%、キャリアでは97.8%のコール率、SMAとキャリアの両方で100%の精度を示しました(表1)。
| 直交法によるコピー数 | 合計 | 一致 | 不一致 | 一致率 | |
|---|---|---|---|---|---|
| SMN1 | 0 | 64 | 64 | 0 | 100.0% |
| 1 | 45 | 44 | 1 | 97.8% | |
| 2 | 897 | 897 | 0 | 100.0% | |
| 3 | 174 | 174 | 0 | 100.0% | |
| 4 | 43 | 43 | 0 | 100.0% | |
| 6 | 1 | 0 | 1 | 0.0% | |
| 合計 | 1224 | 1222 | 2 | 99.8% | |
| SMN2 | 0 | 117 | 117 | 0 | 100.0% |
| 1 | 486 | 465 | 1 | 99.8% | |
| 2 | 541 | 539 | 2 | 99.6% | |
| 3 | 60 | 60 | 0 | 100.0% | |
| 4 | 9 | 8 | 1 | 88.9% | |
| 合計 | 1193 | 1189 | 4 | 99.7% | |
| SMN2∆7-8 | 0 | 1089 | 1089 | 0 | 100.0% |
| 1 | 80 | 80 | 0 | 100.0% | |
| 2 | 4 | 4 | 0 | 100.0% | |
| 合計 | 1173 | 1173 | 0 | 100.0% |
SMN1、SMN2、SMN2∆7-8の集団別コピー数
SMNCopyNumberCallerを1000 Genomes Project(1kGP)の2504の無関係なサンプルとNIHR BioResource Project14の10243の無関係なサンプルに適用し、SMN1およびSMN2コピー数の集団分布を報告しました(図4)。この手法を用いたSMAのキャリア頻度(SMN1のコピーが1つのサンプル)は、過去のPCRベースの調査2、4で報告された頻度と一致しました。特に、SMN1コピー数の変動性は、ほとんどの集団でSMN2コピー数よりもはるかに低く、アフリカ人ではその他の集団よりもSMN1コピー数がはるかに高くなっています。
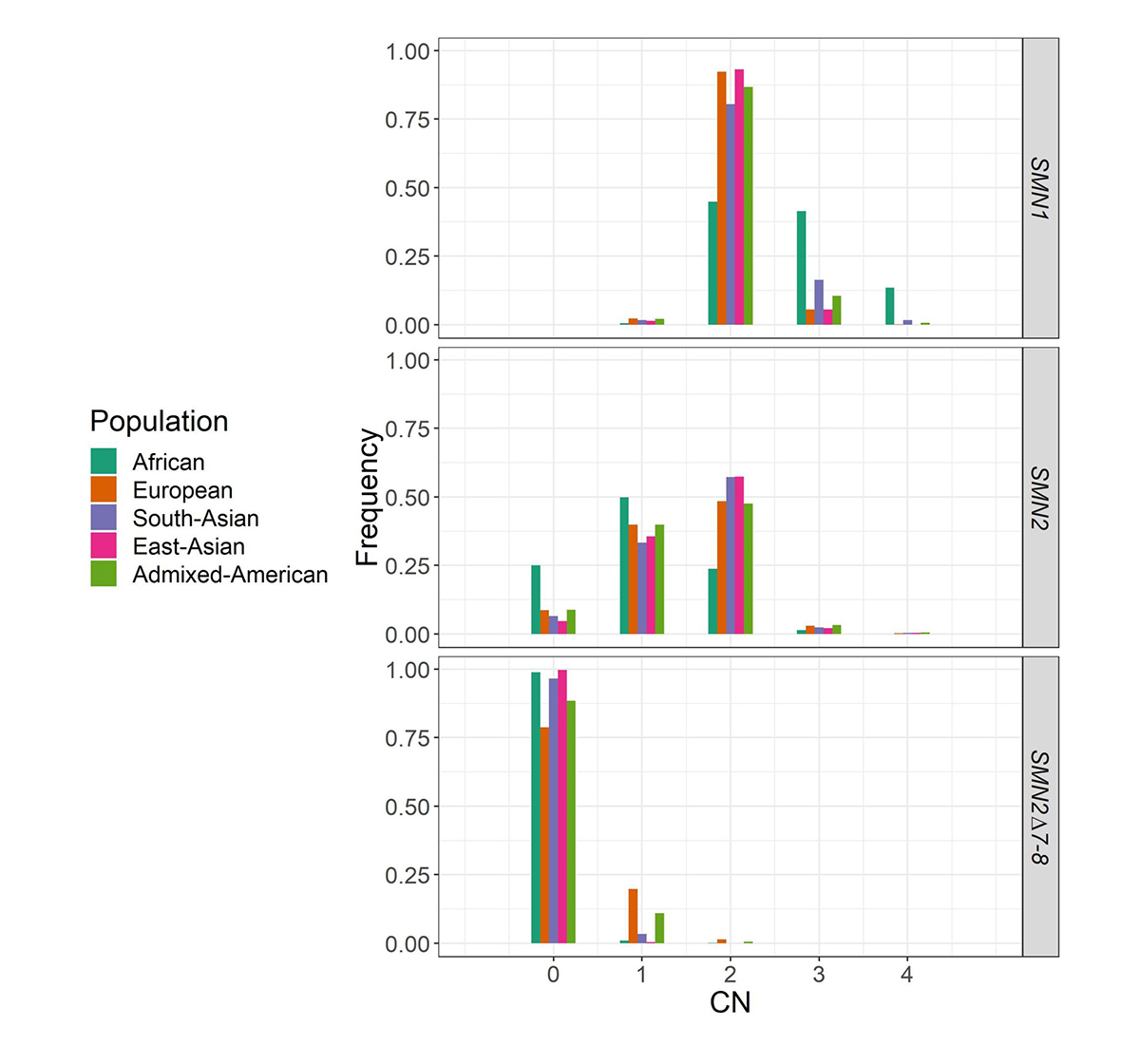
1kGPコホートおよび英国国立健康研究所(NIHR)BioResourceコホートの5つの集団におけるSMN1、SMN2、SMN2Δ7–8のコピー数の分布のヒストグラム。
まとめ
当社のSMNCopyNumberCallerは、SMAのキャリアおよび罹患状態の両方を同定するために使用でき、新生児ケアの包括的な検査およびWGSシーケンスプロジェクトにおける正確なキャリアスクリーニングツールとしてSMA検査を提供できます。ゲノムには、通常のWGSパイプラインではバリアントコールを伝達できない難しい領域がありますが、ここでは、WGSとターゲットインフォマティクスアプローチを組み合わせることにより、このような難解領域の1つを解決する能力を示しています。WGSは、ゲノム全体の遺伝的バリエーションを評価する貴重な機会を提供し、WGSデータを用いて困難な領域に対してよりターゲットを絞ったインフォマティクスソリューションを継続的に開発することで、個別化医療の可能性を現実に一歩近づけることができます。
学術用途向けの詳細情報またはDRAGEN試用版ライセンスについては、dragen-info@illumina.comまでお問い合わせください。
謝辞
我々の共著者であるCambridge 大学のAlba Sanchis-Juan 、Courtney French、 Isabelle Delon、 Lucy Raymondおよび Nemours Alfred I. duPont Hospital for ChildrenのAndrew Connell、 Matthew Butchbach、ならびにイルミナのZoya Kingsbury、Aditi Chawla、Aaron Halpern、Ryan Taft、David Bentleyに感謝します。イルミナのAndrew Warrenが視覚化ツールを開発してくれたことに感謝します。
外部リンク
注釈
- Lunn MR, Wang CH. Spinal muscular atrophy Lancet. 2008; 371(9630):2120–2133.
- Sugarman EA, Nagan N, Zhu H, Akmaev VR, Zhou Z, Rohlfs EM, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72 400 specimens. Eur J Hum Genet. 2012;20(1):27–32.
- MacDonald, WK, Hamilton, D, Kuhle S. SMA carrier testing: a meta-analysis of differences in test performance by ethnic group. Prenat Diagn. 2014;34(12):1219-1226.
- Hendrickson BC, Donohoe C, Akmaev VR, Sugarman EA, Labrousse P, Boguslavskiy L, et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J Med Genet. 2009;46(9):641–644.
- Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388(10063):3017–3026.
- Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-dose gene-replacement therapy for spinal muscular atrophy N Engl J Med. 2017;377(18);1713–1722.
- Rochette CF, Gilbert N, Simard LR. SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum Genet. 2001;108(3):255–266.
- Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96(11):6307–6311.
- Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum Mutat. 2000;15(3):228–237.
- Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10(8):597-609.
- Butchbach ME. Copy number variations in the survival motor neuron genes: implications for spinal muscular atrophy and other neurodegenerative diseases. Front Mol Biosci. 2016;3:7.
- Chen X, Sanchis-Juan A, French CE, Connell AJ, Delon I, Kingsbury Z, et al. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet Med. 2020;22(5):945-953.
- 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526(7571):68-74.
- Turro E, Astle WJ, Megy K, Gräf S, Greene D, Shamardina O, et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature. 2020;583(7814):96-102.