はじめに
タンデムリピート(TR)として知られる比較的短いDNA断片の反復で構成されるシーケンスは、ゲノム全体で起こります(例:図1)。TRの変異率は、他のゲノム領域よりも10~1000倍高いため、TRはヒトの遺伝的変異の大きな一因となっています。TRは、世代間でリピート数が増減するスリッページによって大きく変異します。エビデンスの蓄積により、TRは基本的な細胞プロセス2、3で役割を果たしており、タンデムリピートの大きな伸長は、筋萎縮性側索硬化症(ALS)、脆弱X症候群、およびさまざまな種類の運動失調症などのさまざまな神経障害と関連しています。
CAGモチーフのタンデムリピート。
TRを含む領域のシーケンスでは、リピートシーケンスに部分的または完全にオーバーラップするリードのコレクションが生成されます(図2)。これらのリードのアライメントをつなぎ合わせることで、各ハプロタイプのリピートの長さを決定できます。私たちのグループは、ターゲット4,5とゲノムワイド6のTR解析の両方のためのいくつかの手法を開発しました。ここでは、リード長よりも短いリピートと長いリピートのサイズを推定できる、1つまたは複数の隣接TRを含む領域のターゲット解析のための方法である、ExpansionHunter 4,5に焦点を合わせます。
TRジェノタイピングは非常に困難な問題であり、最善の方法でも、不正確なジェノタイピングコールを生じることがあります。このため、問題のリピートのジェノタイピングに使用されるリードのアライメントを検査するための堅牢な視覚化手法を持つことが重要です。さらに、このような視覚化手法により、臨床的に重要な影響をもたらす可能性のある反復モチーフの変化(例:中断)を検出することができます7,8。標準データ視覚化パイプラインは通常、リファレンスゲノムに対するリードのアライメントの表示に限定されているため、リファレンスに対して拡張されたリピートや異なる長さのアリルを持つリピートには不十分です。これらの問題に対処するために、Repeat Expansion Viewer(REViewer)を開発しました。これは、ExpansionHunterによって再アライメントされたリード出力を視覚化するためのツールです。REViewerは、隣接するリピートをフェージングしてハプロタイプシーケンスを決定し、リードアライメントをこれらのハプロタイプに分配します。結果として得られる静止画像により、特定のジェノタイプコールの精度を視覚的に評価し、リピートシーケンスに中断があるかどうかを同定することができます。
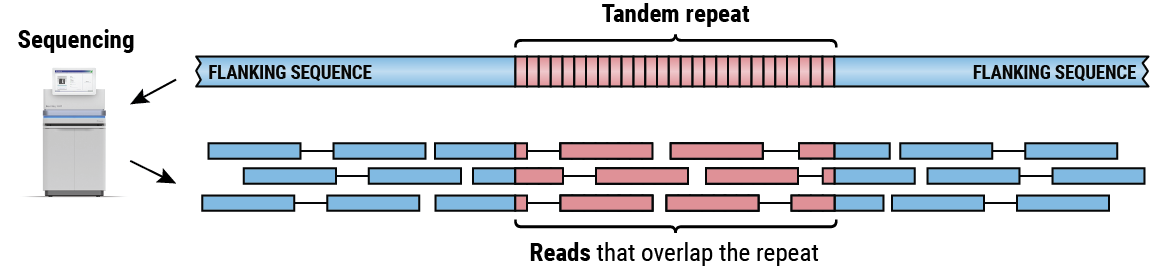
リード長よりも長いタンデムリピートのシーケンスによって生成されたペアリード。
タンデムリピート領域におけるリードのアライメントの視覚化
REViewerは、ExpansionHunterによって生成されたリードのアライメントを表示するように設計されています(図3、ボックス1~3)。これらのアライメントは、ターゲット領域から発生するリードを、そこに位置する1つ以上のリピートをコードする対応するシーケンスグラフに再アライメントすることで得られます5。次に、ReViewerは、ExpansionHunterによって生成されたリピートジェノタイプを使用して推定ハプロタイプシーケンスを構築し、リードアライメントとの一貫性が最も高いハプロタイプペアを選択します(図3、ボックス4~6)。(このステップは、ハプロイド染色体のリピートではスキップされます。) 次に、REViewerは、各ハプロタイプ上の各リードペアの可能なアライメント位置のセットを決定します。例えば、両方のハプロタイプが共有する隣接シーケンス内で発生するリードペアは、各ハプロタイプに正確に1つのアライメント位置を持ち(図3、ボックス7a)、両方のメイトがリピートシーケンスで構成されているリードペアは、十分に長いリピートを持つハプロタイプに複数の可能な起源を持つ(図3、ボックス7b)。リードパイルアップを生成するために、REViewerは各リードペアに対してランダムに1つのアライメント位置を選択します。このステップは、指定された回数(デフォルトでは10,000回)繰り返され、複数のパイルアップを生成します。各ハプロタイプで最も均一なカバレッジを持つパイルアップが視覚化のために選択されます(図3、ボックス8)。
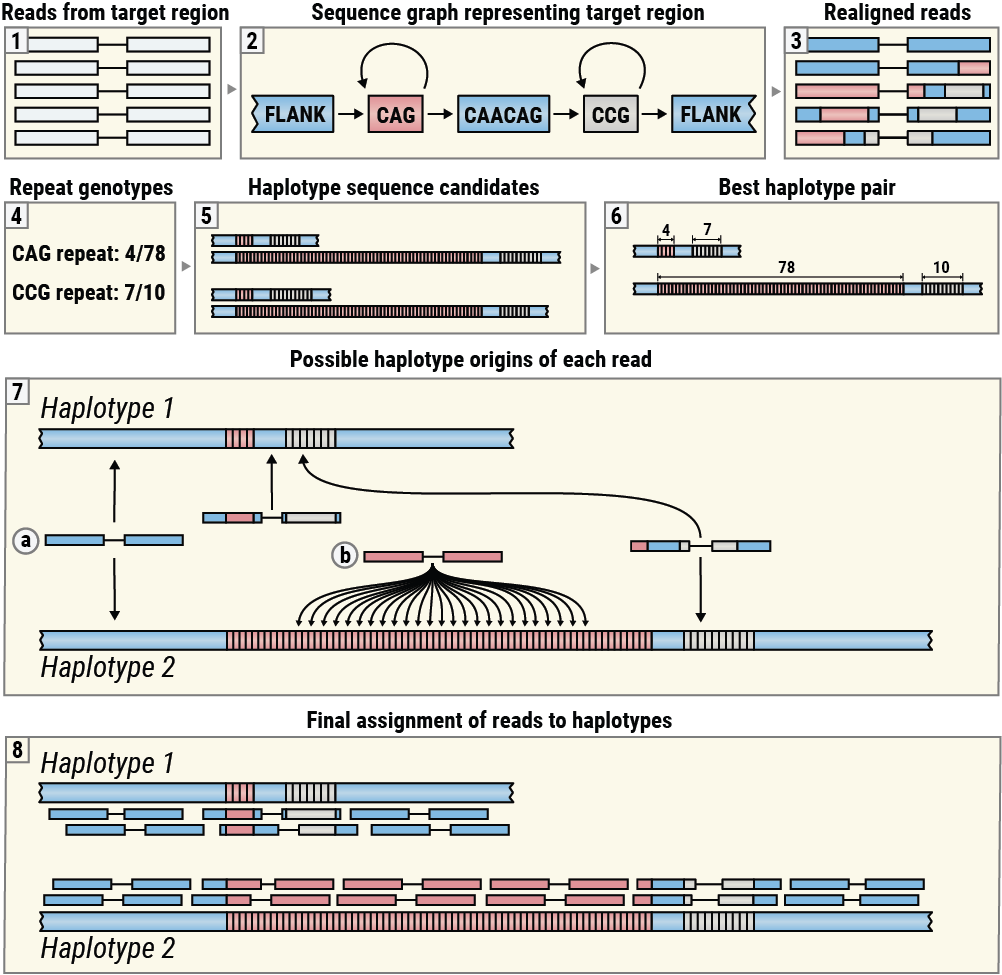
REViewerの視覚化方法の概要。
このアルゴリズムは、特定の遺伝子座のシーケンスが良好で、各成分リピートのジェノタイピングが正しく行われていれば、各ハプロタイプの均一なカバレッジを達成するためにリードを分散させることができるという考えに基づいています。(多くのリードは、特にリピートがホモ接合性であり、結果として生じるハプロタイプが同一である場合、正しい原産地のハプロタイプに割り当てられない場合があります。) 逆に、リピートのサイズが著しく過大評価または過小評価された場合、リードの割り当ては、ジェノタイピングエラーに気づきやすいように均等な山積みにはなりません。
正確にジェノタイピングされたリピートの可視化
この記事の残りの部分では、レビュアーが実際の4およびシミュレーションデータから生成したリードパイルアップの例を確認します。リードによって十分に裏付けられている正確な遺伝子型に対応するパイルアップから始めます。
短いリピート
ATXN3リピートのリードパイルアップについて考えてみましょう。そのアリルは図4に示されているリード長よりも短くなっています。 このリピートは、20/20のジェノタイプ(各アリルに20のモチーフコピー)です。このプロットの各パネルはハプロタイプ(一番上の行に示されているハプロタイプシーケンス)に対応しています。ハプロタイプシーケンスとリードは、リピートシーケンス(オレンジ色)または周囲の隣接シーケンス(青色)とのオーバーラップに従って色分けされます。リードのミスマッチ塩基がすべて表示されます。
パイルアッププロットは、各アリルが多くのスパンリード(リピート全体に及ぶリード)によってサポートされており、アライメントに不一致のあるリードがないため、遺伝子型コールがリードによって十分にサポートされていることを示しています。(アライメントの不一致は、リードが2つのハプロタイプのいずれかと一致しないことを意味します。例えば、40リピートのリードは遺伝子型20/20と一致しないことになります。) リピートシーケンスに明らかな中断の証拠があります。例えば、3番目から最後のモチーフのシトシンはチミンに変異します。
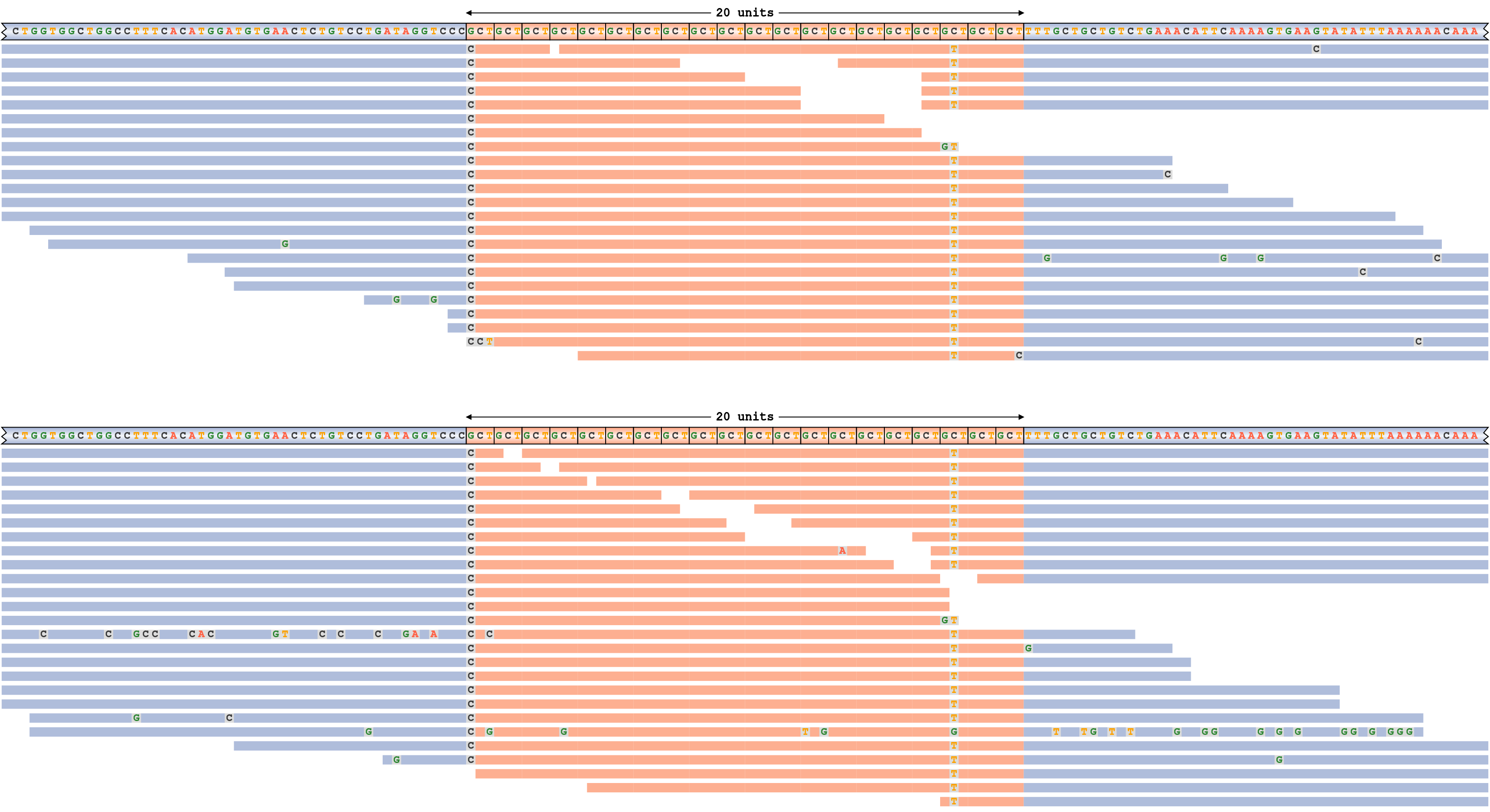
ジェノタイプ20/20のATXN3リピートのリードパイルアップ。シーケンスの中断は、ほとんどのリードアライメントでミスマッチがある位置に対応します。
拡張リピート
図5は、拡大したアリルを有するDMPKリピートを示す。REViewerは、リピート全体にリードを分散させ、ハプロタイプ全体に同様のリードカバレッジを達成できたため、拡張リピートはリードによって十分にサポートされています。(リピート内のリードのアライメント位置はランダムに選択されることを覚えておくことが重要です。) 短いアリルは、多数のスパンリードでも十分にサポートされています。淡い色で描かれたアライメントは、いずれかのアリルに割り当てることができるリードに対応しています。
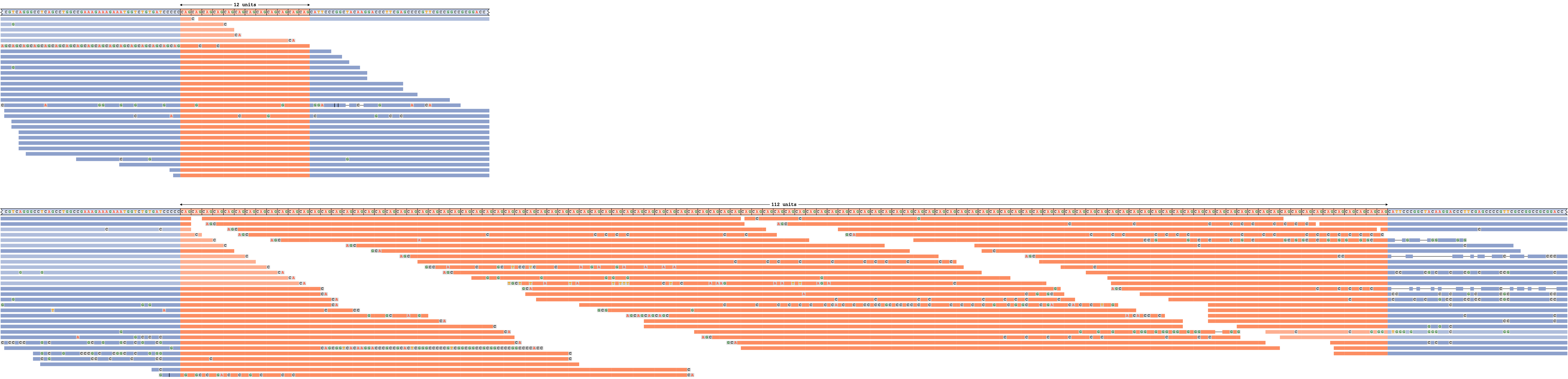
DMPKリピートのリードパイルアップで、1つのアリルに伸長します。
2つの隣接するリピートを持つ遺伝子座
REViewerのより複雑な適用を実証するために、病原性CAGリピートと付近の“迷惑”CCGリピートの2つの隣接するリピートを含むHTTリピート領域に適用しました。前者のリピートは14/17、後者のリピートは9/12と遺伝子型が決定されます。したがって、図6に示すハプロタイプの1つはサイズ14と12のリピートをそれぞれ含み、他のハプロタイプの1つはサイズ17と9のリピートを含みます。両方のハプロタイプがリードによって十分に裏付けられていることは明らかです。さらに、パイルアッププロットは、両方のハプロタイプでCCG反復モチーフの2番目のコピーにGからAへの変異があることを示しています。
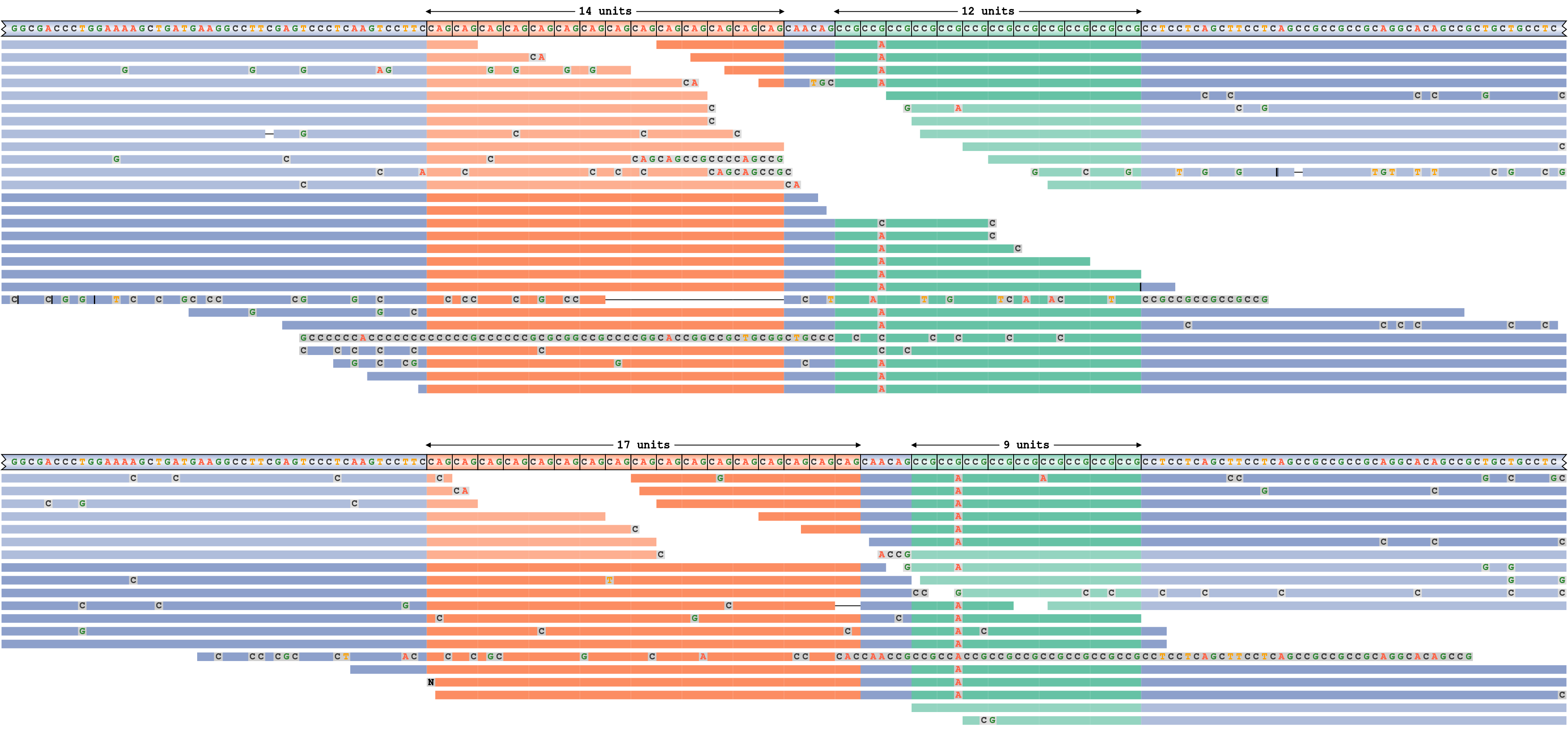
近くの2つのリピートを含むHTT遺伝子座のリードパイルアップ。
不正確なジェノタイピングリピートの可視化
このセクションでは、不正確なジェノタイピングリピートに対応するリードパイルアップの例について説明します。偽陽性と偽陰性の反復拡張コールの両方を例示するために、シミュレーションデータを使用します。
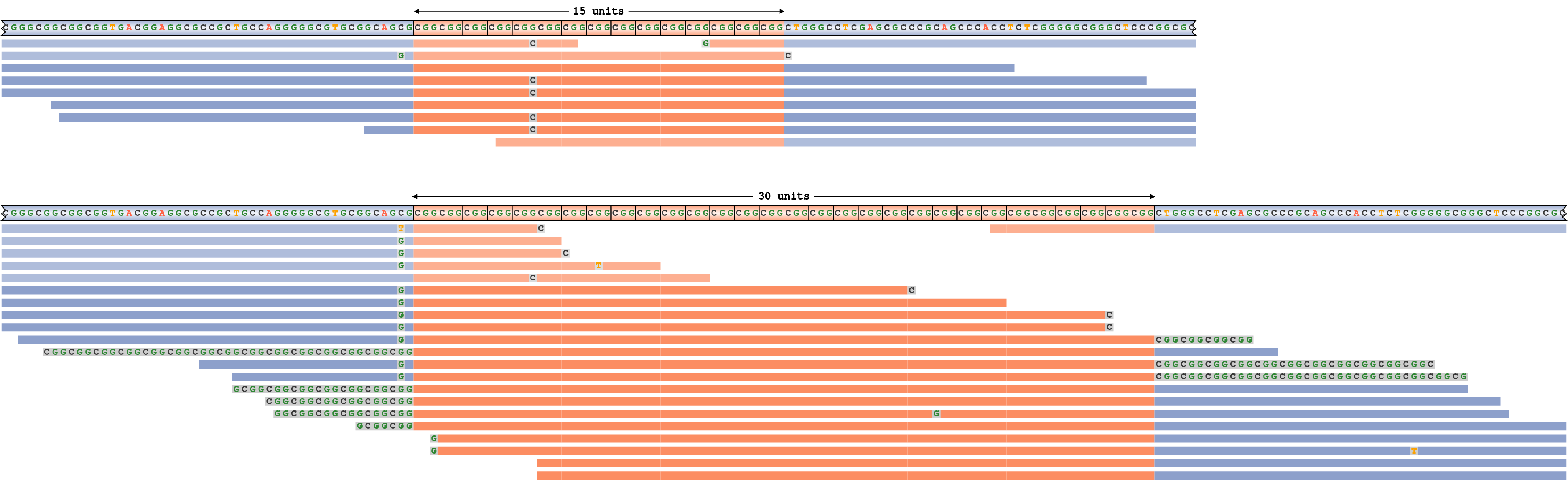
過大評価されたリピートサイズ
偽陽性リピート伸長コールに対応するパイルアップの例を示すために、ホモ接合性ジェノタイプ10/10のC9ORF72リピート領域からのリードをシミュレーションしました。次に、CC9ORF72リピートシーケンスにやや近いほぼ完全なCホモポリマーリードをスパイクし、リピート遺伝子型を10/10ではなく10/30に強制的に実行しました。図7は、対応するリードのパイルアップを示しています。予想通り、この山は、より長いリピートを持つハプロタイプに配置されたリードの1つを除くすべては、より短いハプロタイプと一致しており(これらのリードは淡い色で示されています)、1つのアライメントが不十分なリードのみが拡張をサポートすることを示しています。実際には、これは1回の低品質リードによって引き起こされる偽陽性コールであると考えられます。
CC9ORF72リピートの伸長と誤って呼ばれた。
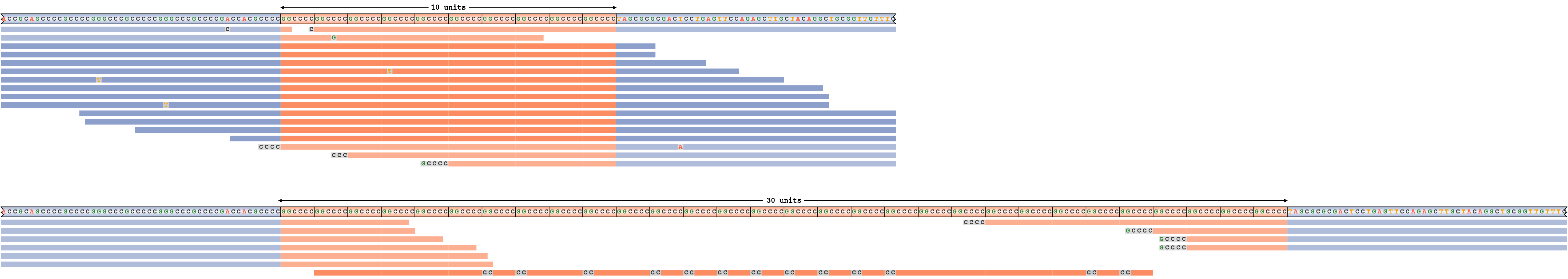
過小評価されたリピートサイズ
偽陰性リピート伸長コールの例を生成するために、ジェノタイプ15/55のFMR1リピートをシミュレーションし、次にREViewerに(不正確な)ジェノタイプ15/30に対応するリードパイルアップを生成するよう強制しました。図8は、結果として生じるパイルアップを示しています。サイズ55のリピート内のリードを照合するために、REViewerはアライメントの端を最長のアリルのサイズにクリップしました。リードの実際のクリップ部分は、ベースシーケンスを示すグレーのセグメントとして表示されます。リピートと30のモチーフがオーバーラップするリードが過剰であり、これらのリードはすべてリピートシーケンスで構成されているため、リピートのサイズは過小評価されている可能性が高いと結論付けています。
最長のアリルのサイズが過小評価されている遺伝子型に対応するFMR1リピートパイルアップ。
制限事項
REViewerは、ExpansionHunterによって生成されたリピートジェノタイプとのシーケンスデータの一貫性を評価するツールです。臨床現場でのジェノタイプコールを裏付けるエビデンスをレビューし、問題のあるコーナーケースを特定して将来の開発を促進するメカニズムを提供します。REViewerによって生成されたリードパイルアッププロットには、不正確性が含まれている可能性があります。リピートのフェーズが正しく行われない場合(リピートが互いに離れている場合など)があり、両方のハプロタイプと一致するリードペアが間違ったハプロタイプに割り当てられることがよくあります。また、REViewerの最新バージョンでは、スパンが断片長を超えないリピートを視覚化します(長いリピートは断片長で上限が設定されます)。
結論
我々は、ExpansionHunterによって決定されたリピートジェノタイプをサポートするリードのアライメントを視覚化するためのツールを開発しました。全長アライメントを表示するために、REViewerは、互いに近接して位置するすべてのリピートの遺伝子型をフェージングすることにより、ターゲット座位のハプロタイプ配列を決定します。REViewerは、ハプロタイプ間およびハプロタイプ内の両方でリードができるだけ均等に拡散するように、複数の起源を持つリードの位置をランダムに選択しながら、ハプロタイプ間でリードを分散します。多くの個別のリードの配置は正しくないかもしれませんが、プロットにより、特定のジェノタイプコールの効率的な視覚的評価を行うことができます。また、これらのプロットは、リピートシーケンスおよびリピートのすぐ周辺のシーケンスの中断を検出するために使用できることも実証しました。その存在が臨床的意味を持つ可能性のあるシーケンスを中断させる能力を開発し、検証する作業が進行中です。
最後に、REViewerは短いインデルを視覚化するために使用でき(ExpansionHunterはこのバリアントタイプに対して基本的なサポートを持っているため)、原則として他のバリアントタイプと連携するように作成できる点に注意してください。REViewerの使用に関して問題が生じた場合や、プログラムの改善についてご提案がある場合は、お気軽にお問い合わせください(Egor edolzhenko@illumina.comおよびMike meberle@illumina.com)。
外部リンク
- ExpansionHunter:https://github.com/Illumina/ExpansionHunter
- REViewer:https://github.com/Illumina/REViewer
参考文献
- Fan H, Chu JY. 短いタンデムリピート変異の簡単なレビュー。 ゲノミクスプロテオミクスバイオインフォマティクス。2007年2月;5(1):7-14. doi: 10.1016/S1672-0229(07)60009-6。
- Gymrek M, Willems T, Guilmatre A, et al. ヒトにおける遺伝子発現変動に対する短いタンデムリピートの豊富な寄与。 Nat Genet. 2016年1月;48(1):22-9。
- Hannan AJ. タンデムリピートは、健康と疾患における遺伝的可塑性を媒介します。 Nat Rev Genet. 2018 May;19(5):286-298。
- Dolzhenko E, van Vugt JJFA, Shaw RJ, et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. ゲノム樹脂 2017 Nov;27(11):1895-1903。
- Dolzhenko E, Deshpande V, Schlesinger F, et al. ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. バイオインフォマティクス。 2019年11月1日;35(22):4754-4756。
- Dolzhenko E, Bennett MF, Richmond PA, et al. ExpansionHunter Denovo:ショートリードシーケンスデータにおける既知および新規のリピート伸長を見つけるための計算方法。 ゲノムバイオル。2020年4月28日;21(1):102。
- Kraus-Perrotta C, Lagalwar S. 拡大、モザイク、中断:脊髄小脳失調症1型におけるCAG反復変異のメカニズム。 小脳性運動失調症。2016年11月22日、3:20。
- Wright GEB, Collins JA, Kay C, et al. ポリグルタミンのサイズとは無関係の、中断のないCAGの長さは、体細胞不安定性の増加、ハンチントン病の発症の加速をもたらします。 J Hum Genetです。 2019年6月6日;104(6):1116-1126。