DRAGEN STRに関する重要なポイント
- DRAGEN DNAパイプラインには短鎖タンデムリピートコーラーが含まれています
- PCRフリーの全ゲノムシーケンスと組み合わせることで、このコーラーは、FMR1、ATXN1、HTT、C29orf72などの遺伝子における医学的に関連のあるリピート伸長に対して98%を超える感度を示します。
- 偽陽性は稀(< 1%)ですが、推定陽性について確認し、より精確に判定するために確認研究を行うことをお勧めします。
STRとは何ですか?なぜ重要なのですか?
ショートタンデムリピート(STR)は、DNAのシンプルなシーケンスが連続してコピーされるゲノム領域です(表1)。ヒトゲノムには多くのSTR領域があり、そのほとんどは既知の機能を持っていません。
| A氏:(CAT)×3:...CATCATCAT... |
| B氏:(CAT)×1:...CAT... |
| C氏:(CAT)×9:...CATCATCATCATCATCATCATCATCAT... |
表1 STRに関する仮説例
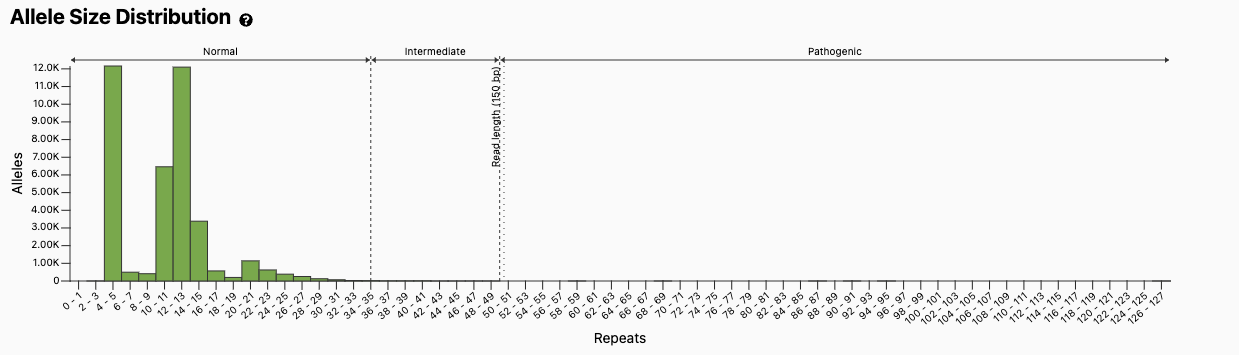
図はgnomAD v3データベース、PMID 32461654から抜粋1
STR変動と医学的疾患との関連性が初めて発見されたのはハンチントン病でした。Marcy MacDonald, PhDと彼女のチームの画期的な探索の歴史についての詳細は、Nature Educationのこちらの記事をご覧ください。 ハンチントン病患者は、疾患名(HTT)にちなんで名付けられた遺伝子に3つのヌクレオチドC-A-G(「トリグリセリドリピート」)が連続40セットを超えて存在しています。この配列は遺伝子のコード配列内であり、アミノ酸グルタミンのリピートに翻訳されます。結果として生じるタンパク質中の連続したグルタミンの数が増加すると、ニューロンに凝集が起こり、最終的には運動失調や神経学的低下などのハンチントン病の臨床徴候や症状を引き起こします。
常染色体優性遺伝パターンを持つハンチントン病の家族では、罹患した人の子供は、症状の発症が早く、より急速な神経変性経過をたどることが多いことが指摘されました。STR解析では、リピートの伸長は重度の影響を受けた子孫でさらに伸長していつことが示され、この悲劇的な現象のメカニズムを提供しました。現在、この現象は「表現促進現象」と呼ばれています。
ハンチントン病の最も狡猾な側面の1つは、一般的に患者が子供を持つ頃までその症状が現れず、その結果、子供に伸長したSTRが遺伝するリスクは50%になることです。現在利用可能なツールを使えば、技術的には、この疾患の全数スクリーニングをはるかに早い時点で行うことができます。しかし残念なことに、有効な治療法がまだないため、一般集団にこの種の検査を推奨することは適切ではありません。ハンチントン病のターゲット遺伝子治療を追求する取り組みが進行中です。ハンチントン病の遺伝子ベースの治療法が開発できれば、すべての人を対象にした検査が正当化されるかも知れません。
HTTリピート伸長の発見は、他の疾患でこのタイプのバリアントを探索している他のグループを鼓舞しました。STRがヒト疾患と関連している遺伝子は、現在のところ56種類以上あります(図2)。これには、遺伝性知的障害の最も一般的形態の1つであり、米国臨床遺伝・ゲノム学会がキャリアスクリーニングに推奨する最上位の病態である脆弱X症候群(FMR1 gene)が含まれます。
図はgnomAD v3データベース、PMID 32461654から抜粋1
STRは通常どのように解析されますか?
STRを評価するための最初の効果的な手法は、サザンブロットでした。この手法は高感度ですが、ラボでの実施は煩雑であり、精確なリピート数を評価することが困難です(図3)。
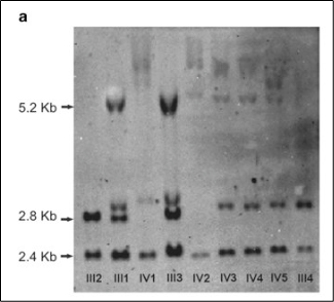
PMID 21107340から抜粋した切り抜き図。2 原文のキャプション:「FMR1(脆弱X精神遅滞1)遺伝子のサザンブロット解析。正常非メチル化(2.8 kb)、正常メチル化(5.2 kb)およびコントロールバンド(2.4 kb)のサイズを示す。
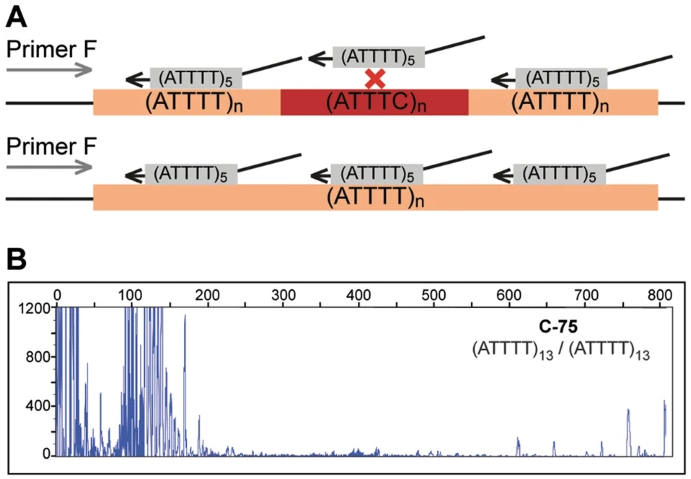
PMID29891931から抜粋した切り抜き図。3原文のキャプション:「DAB1で大きな五塩基アレルを検出するためのATTTT RP-PCR。a反復的なATTTT領域とアニーリングし、正常アレルおよび変異アレルにおけるDNA増幅をもたらすATTTT RP-PCRプライマーの概略図。b表1の対照個体における蛍光ATTTT RP-PCR解析を示す電気泳動図:C-75、C-88、C-91、C-95、およびC-44:およびSCA37の罹患個体A-1およびA-9」
STRはパネルデータまたはエクソームデータを使用して解析できますか?
残念なことに、特定の繰り返しパターンでは、NGSを用いたパネルシーケンスやエクソームシーケンスに必要なライブラリー調製やターゲット増幅またはハイブリダイゼーションプロセスは、アッセイから反復DNAを除去できます。最大限のバイオインフォマティクスを用いても、チューブに存在しないシグナルをすくいあげることはできません。とはいえ、エンリッチメントベースの次世代シーケンサーは特定の座位に対して実行可能であり、エクソームベースの検査の診断率を向上させることができると、最近の研究では示されています。プローブキャプチャーとPCR増幅の効果は、すべてのSTRパターンで同じではありません。この現象は、既知のすべてのSTR座位で完全には特徴付けられたわけではありませんが、100% GCモチーフは特に増幅が困難であることが全般的に理解されています。
DRAGEN STRはどこに使用されていますか?
パネルシーケンスやエクソームシーケンスとは対照的に、PCRフリー全ゲノムシーケンス(pfWGS)はシーケンス用の反復ゲノムDNAを保持します。研究者にとっての課題は、最も関連性の高い伸長アレルが、イルミナの短鎖リードシーケンスデータのリード長を超えることが多い場合のリピート長のジェノタイピングです。この問題に対処するために、イルミナの卒業生Egor Dolzhenko, PhD、Michael Eberle, PhDと同僚たちはExpansionHunterを開発しました。まず、彼らは被験者データを比較できる重要なSTR領域のためにカスタムリファレンスセットを作成しました。このアルゴリズムは、隣接領域におけるリードや、ペアエンドメイトとともにリピート配列を含むリードなど、pfWGSデータに情報を与えるシーケンスリードを同定します。特別に準備されたリファレンスをこれらのリードと組み合わせることで、このアルゴリズムは非伸長アレルを容易に同定し、伸長が疑われるアレルにはフラグを付けることができます。
Illumina DRAGEN secondary analysis には、pfWGSデータを持つサンプルのオプションとしてExpansionHunterを使用したSTRジェノタイピングが含まれています。これは、ローカルハードウェアでもクラウド上でも実行できます。保有されているデータにDRAGENワークフローを実装することに興味がある場合は、ぜひお問い合わせください!
数学やバイオインフォマティクスに興味のある方にとって、一次文献は優れたリソースです。過去の論文でさらに詳しく説明しています。
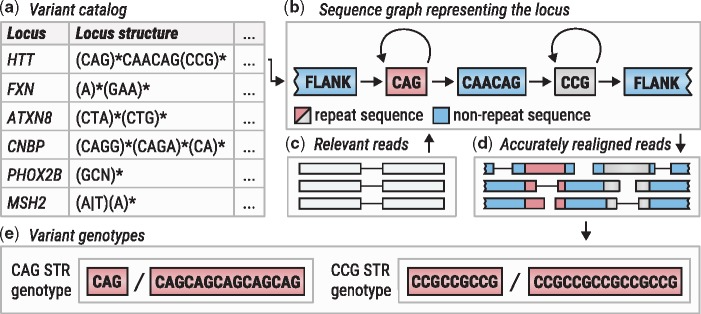
PMID31134279から抜粋した図。4原文のキャプション:「ExpansionHunterの概要。(a) バリアントカタログファイルから座位定義が読み取られる。(b) バリアントカタログの仕様に沿ってシーケンスグラフが作成される。(c) 関心リードをインプットバイナリアライメント/マップファイルから抽出される。(d) リードをグラフにアライメントする。(e) アライメントは各バリアントをジェノタイピングするためにつなぎ合わされる。」
DRAGEN STRの仕組み
複数のSTR拡張条件に対応する一連の陽性および陰性データセットに直面した場合、ExpansionHunterは優れた性能を示しました(図6)。1つを除き、陽性対照はすべて陽性で、非常に高い精度を示しました。陰性の予測値(真陰性検査の陰性の割合)も非常に高い精度でしたが、正常対照は陽性という結果になりました。これらの結果が組み合わさることで、すべての参加者に対しpfWGSを実施した後、rpPCRで追跡調査を行って、参加者の値がラボで検証されたカットオフ値を超えた遺伝子の潜在的な伸長を確認できる、「スクリーニングと確認」のアプローチが強く支持されました。
図6の研究における唯一の偽陰性は、さらに考察する価値があります。このサンプルにはFMR1に前変異がありますが、既定のカットオフ値では正常と判定されました。このことから、臨床検査室は、この伸長タイプに対する感度を最大限に確保するためには、わずかに低いカットオフ値を使用し、多少高いrpPCR確認率を受け入れることを検討することが提案される。これは、感度と特異性のバランスを取るというギブアンドテイクの典型的な例でしょう。
全ゲノムシーケンス用臨床検査機器の開発の一環として、157のアレルが11の座位にわたって臨床的な関連性を持って拡大し、STR座位あたり約700の既知の陰性アレルが同様の方法で評価されました。この研究では、感度は98%で、サンプルあたりの偽陽性率は1%未満でした。ATXN1およびATXN2に対して、より許容性の高いカットオフを使用した場合、感度は100%に改善されました。
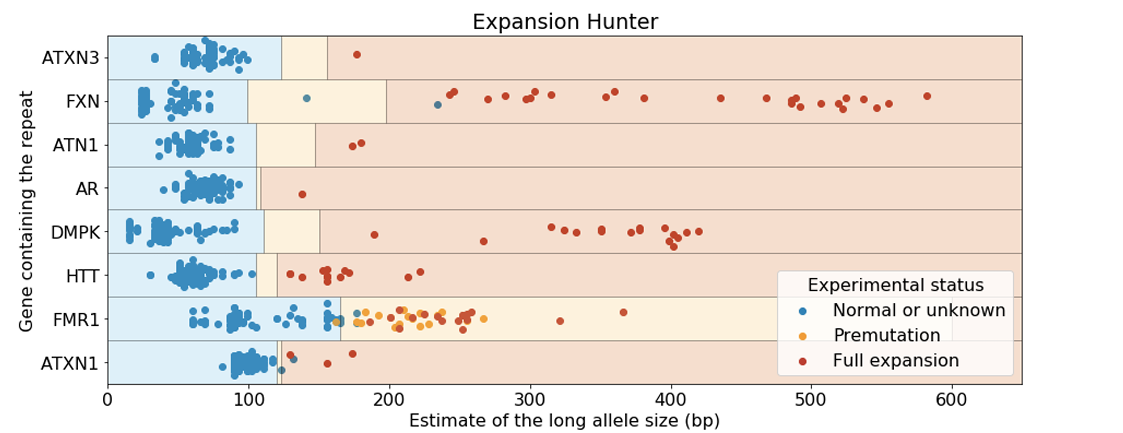
PMID 31134279から抜粋した切り抜き図。4原文のキャプション:「既知のリピート伸長を有するCoriellサンプルの解析。青色、オレンジ色、および赤色の四角形は、対応するリピートの正常、前変異、および完全伸長の予測サイズの範囲をそれぞれ定義する。各ドットは最長のアレルのサイズに対応しており、その色は実験的に定義されたステータスに従って設定した。GangSTRは、既定のオフターゲット座位が提供されたSTRでのみ実行した。GangSTR値は、FMR1を除くすべての遺伝子に対して『ゲノムワイド』モードを使用して算出し、FMR1は、このリピートではるかに優れた性能を示したターゲットモードを使用して解析を行った。リピートサイズは600 bpを上限とした。
この研究は、英国を拠点とする100,000 Genomes Projectチームによって再現され、その集団規模のデータセットにおけるSTRの評価が行われました(PMID: 35182509)。5この研究では、拡張障害と一致する神経学的表現型を持つ404人の個人から、潜在的なSTR拡張がWGSとExpansionHunterによって同定されました(図7)。PCR検査により陽性の可能性が確認されました。彼らの調査結果は印象的でした。
「全ゲノムシーケンスでは、221の伸長アレルのうち215、1321の非伸長アレルのうち1316が正しく分類され、PCR検査結果と比較した場合、13の疾患関連座位全体で97·3%の感度(95% CI 94·2~99·0)と99·6%の特異性(99·1~99·9)が示されました」
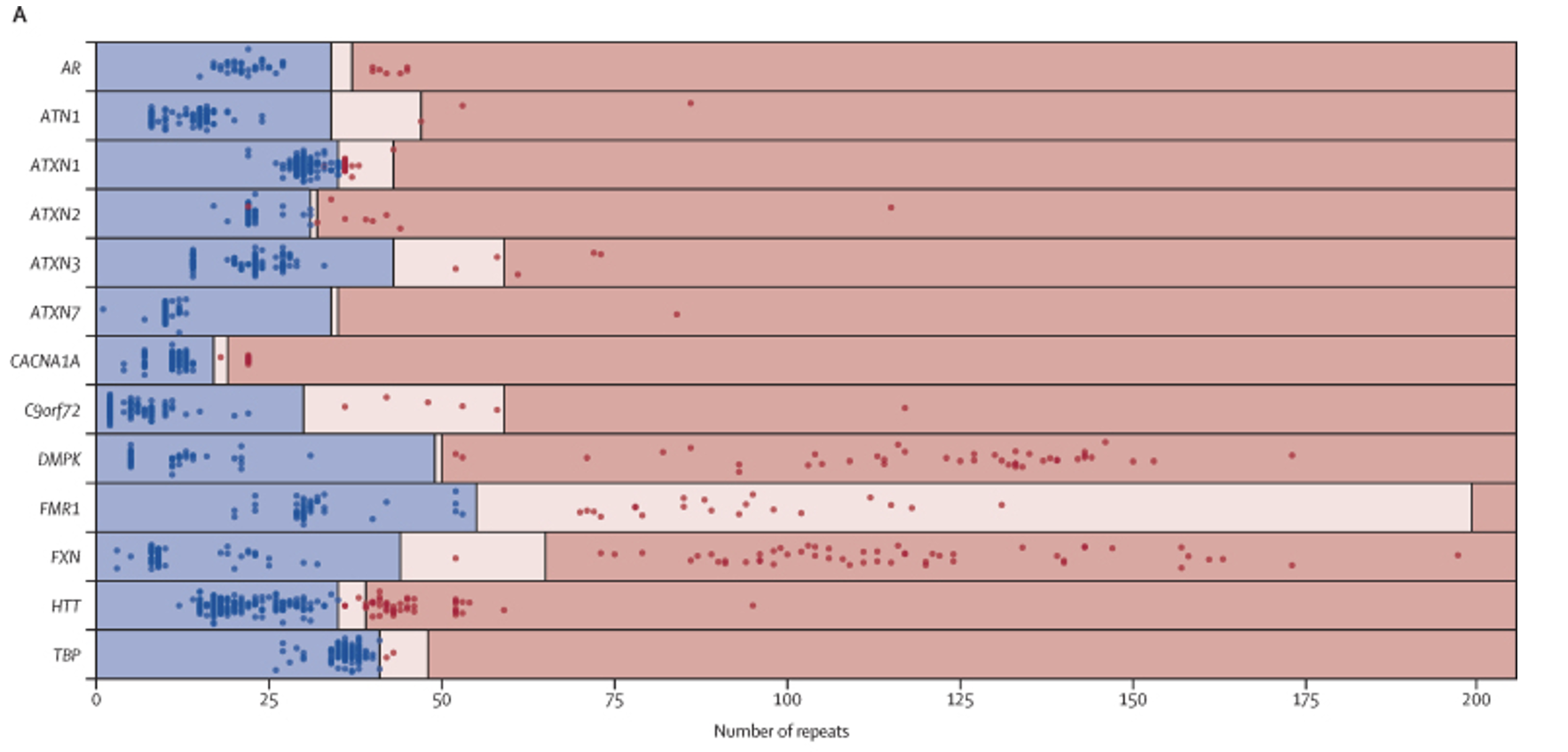
PMID 35182509から抜粋した切り抜き図。5原文のキャプション:「全ゲノムシーケンスを使用したリピート伸長検出の性能:793の伸長コールにわたって拡張ハンターによって予測されるリピート伸長のサイズを示すスイムレーンプロット。各ゲノムは2つの点で表されます。1つは各座位の各アレルに対応しますが、男性のX 染色体(すなわちFMR1とAR)上のものを除き、1つの点のみが表示されます。点は目視点検後に拡張ハンターによって推定されたリピート長を示し、色はPCRによって評価されたリピートサイズを示します(青色は非拡張、赤色は拡張を示します)。領域は、付録(p 28)に示すように、各遺伝子の非拡大(青色)、前変異(ピンク色)、および拡大(赤色)の範囲を示すように網掛けされています。ピンク色または赤色の網掛け領域にある青色の点は偽陽性を示し、青色の網掛け領域にある赤色の点は偽陰性を示します。個別の電話連絡は、付録(p 27)に記載されています。
新しいSTR座位を発見することは可能ですか?
対象疾患の範囲を大幅に改善し、未診断の疾患の解決を目指す研究者を支援するために、拡張ハンターチームは、ゲノムを精査して反復リードの山を見つけ、影響を受けた個人と対照サンプルのコホート間でこれらの山のカバレッジと位置を比較することで、推定STRの拡張を同定するための新しいアプローチを考案しました。ExpansionHunter Denovoと呼ばれるこの新しいツールを用いて、フリードライヒ運動失調症や脆弱X症候群などの古典的なSTR疾患が「再発見」されました(図8)。全体として、このアプローチを用いて、既知の44伸長のうち41伸長が陽性と確認されました。このパッケージのリエンジニアリング版は、今後のDRAGENリリースが予定されています。詳細については、お問い合わせください。
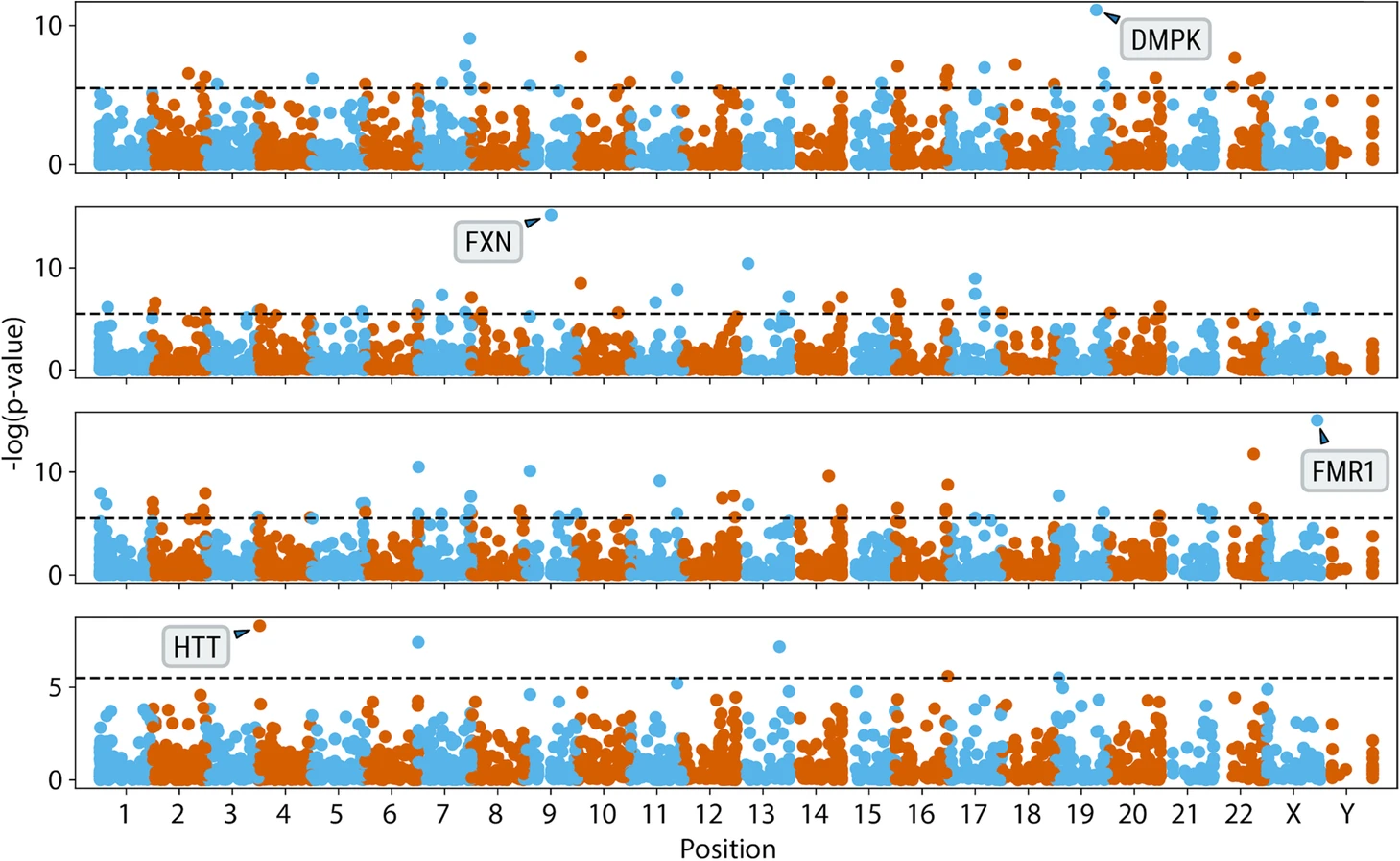
PMID 32345345から抜粋した図。5原文のキャプション:「DMPK、FXN、FMR1、HTT遺伝子(上から下へ)の病原性伸長が判明している症例を150の対照と比較するアンカーIRRのゲノムワイド解析」
STR検出はどのように実行できますか?
ExpansionHunterを実行できる2つの主な方法は、DRAGENワークフローの一部として、またはスタンドアロンのソフトウェアとして独立して実行することです。DRAGEN DNAseqパイプライン3.7.5以降のバージョンには、ExpansionHunter解析を実行するオプションが含まれます(詳細はオンラインヘルプを参照)。DRAGENは、物理ハードウェア(「オンプレミス」)または複数のプラットフォーム上でクラウドベースのワークフローの一部として実行できます。このソフトウェアは、GitHub上でスタンドアロンのパッケージとしても利用できます(表2)。
| プラットフォーム | タイプ | 説明 | リンク | ||||||
| DRAGEN二次解析 | オンプレミスサーバー* | 二次ゲノム解析(アライメントおよびバリアントコール)の精度と速度が最適化されたカスタムデザインのコンピューターハードウェア。 | https://www.illumina.com/products/by-type/informatics-products/dragen-bio-it-platform.html | ||||||
| Emedgene | クラウド | パネル、エクソーム、全ゲノムを含む、ゲノム解析のためのクラウドベースのプラットフォーム。これには、DRAGEN二次解析、アノテーション、フィルタリング、三次解析のワークフロー、知識データベース、ロバストなレポート作成ツール、および人工知能ベースのバリアントへの優先順位付けが含まれます。 | |||||||
| BaseSpace Sequencing Hub | クラウド | イルミナのシーケンスランと解析を管理するために設計されたクラウドベースのバイオインフォマティクスプラットフォーム。 | |||||||
| Illumina Connected Analytics | クラウド | さまざまなプロジェクトや種類にわたるデータ管理と解析を行うよう設計されたクラウドベースのバイオインフォマティクスプラットフォーム。 | https://www.illumina.com/products/by-type/informatics-products/connected-analytics.html |
||||||
| TruSight Software Suite | クラウド | エクソームと全ゲノムをエンドツーエンドでゲノム解析するクラウドベースのプラットフォーム。 | https://www.illumina.com/products/by-type/informatics-products/trusight-software-suite.html | ||||||
| Linux | ソフトウェア | オリジナルのExpansionHunterソフトウェアパッケージが研究用に用意されており、お客様のサーバーで実行できます。 |
*オンプレミスとは、サーバールーム/キャビネットにインストールされている物理的なオンプレミスコンピューターハードウェアを指します。
表2
学術用途向けの詳細情報またはDRAGEN試用版ライセンスについては、dragen-info@illumina.comまでお問い合わせください。
2024年6月4日:この記事は、重要なポイントに関する新しいセクション、「STRをパネルデータまたはエクソームデータで解析することは可能か?」、「DRAGEN STRの性能はどうか?」、「新しいSTR座位を発見することは可能か?」、というセクション内の新情報、および新しい図7によりアップデートされました。
注釈
- Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434-443. doi:10.1038/s41586-020-2308-7
- Martorell L, Nascimento M, Colome R, Genovés J, Naudó M, Nascimento A. Four sisters compound heterozygotes for the pre- and full mutation in fragile X syndrome and a complete inactivation of X-functional chromosome: implications for genetic counseling. J Hum Genet. 2011;56:87-90. doi:10.1038/jhg.2010.140
- Loureiro JR, Oliveira CL, Sequeiros J, Silveira I. A repeat-primed PCR assay for pentanucleotide repeat alleles in spinocerebellar ataxia type 37. J Hum Genet. 2018;63:981-987. doi:10.1038/s10038-018-0474-3
- Dolzhenko E, Deshpande V, Schlesinger F, et al. ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics. 2019;35(22):4754-4756. doi:10.1093/bioinformatics/btz431
- Ibañez K, Polke J, Hagelstrom RT, et al. Whole genome sequencing for the diagnosis of neurological repeat expansion disorders in the UK: a retrospective diagnostic accuracy and prospective clinical validation study. Lancet Neurol. 2022;21(3):234-235. doi:10.1016/S1474-4422(21)00462-2
- Dolzhenko E, Bennett MF, Richmond PA, et al. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol. 2020;21:102. doi:10.1186/s13059-020-02017-z