precisionFDA Truth Challenge V2とCMRGとは何ですか?また、より良いゲノムを提供する上でどのように役立ちますか?
イルミナで開発された全ゲノムシーケンス(WGS)テクノロジーとソフトウェアを使用すると、ヒトゲノム60億ヌクレオチドのほぼすべてを高い精度と効率で解析できます。これらの進歩は、私たちの細胞がどのように機能するかの奥深い基礎研究から、特定の変異をターゲットとするプレシジョンメディシン療法まで、ヘルスケアにおけるゲノミクス革命を推進しています。
PrecisionFDA Truth Challenge V21は、最高水準の小規模バリアントコールを評価するように設計されており、3つのサンプル(HG002、HG003、HG004)のゲノム全体の92%をカバーするベンチマーキングセットを生み出しました。Illumina DRAGENのマッパーとバリアントコーラーは、ママッピング困難領域と全ベンチマーク領域のカテゴリーにおいて、イルミナのリードに最も高い精度をもたらしました。さらに、2番目に優秀な競争相手よりもコールエラーがそれぞれ38%および28%少ないという結果でした。
precisionFDAチャレンジを通じて発表されたベンチマーキングコールセットでは、マッピング困難領域、セグメント重複、および主要組織適合性複合体(MHC)領域など、技術的に困難な領域における精度の評価に焦点を当てていましたが、ヒトゲノムのその他の領域も同等か、もしくはさらに困難になる可能性があることに留意することが重要です。一部のバリアントと遺伝子は、標準的な解析方法に抵抗性があり、より深い探索を必要とします。全ゲノムシーケンスをかつてないほど向上させる努力を重ねる中で、これらは私たちが最重要視する問題となっています。ハンチントン病2のような疾患を引き起こすショートタンデムリピート伸長を評価する方法や、パーキンソン病やゴーシェ病のリスクに関与するGBA1遺伝子の解明など、これまでのブログ記事でいくつかの研究を取り上げました。3
ヒトの健康にとって最も重要なヒトゲノムの技術的に困難な領域におけるベンチマーキングを強化するために、米国立標準技術研究所(NIST)とWGSを実施するトップ機関の共同研究者は、方法論全体で一貫した比較点として使用されることを目的として「医学的に重要」と「技術的に困難な」という2つの概念の交差点である「困難な医学的に重要な遺伝子」(CMRG)のリストを公開しました4。このようにして、CMRGは、マッピングが難しいという技術的概念を拡大し、遺伝性疾患に関連するバリアントを持つ領域に焦点を絞っています。
医学的に重要なリストには、ClinGenによってキュレーションされた遺伝子、返却する二次的所見としてAmerican College of Medical Geneticsによって推奨された遺伝子、Clinical Pharmacogenetics Implementation Consortiumのファーマコゲノミクス遺伝子、および米国産科婦人科学会によって推奨されたキャリアスクリーニングのためのコア遺伝子が含まれます。これらは幅広い医療への適用にまたがる遺伝子であり、生涯にわたって人の健康に多大な影響を与える可能性があります。
「困難な領域」は、Genome in a Bottle Consortiumによって、十分に特徴付けられた細胞株サンプルで標準解析を行う際に、偽陰性および/または偽陽性の結果が観察される領域と定義されています。
precisionFDA Truth Challenge V2とCMRGは、イルミナのWGSデータから精度を測定するデータセットをイルミナに提供し、かつてはアクセス不可能と考えられていた領域の精度を実証する機会を提供しました。これらの機会は、イルミナの実績ある高精度シーケンスとDRAGENの革新的なインフォマティクスを組み合わせ、ゲノム全体で高精度のバリアントコールを提供することの利点を浮き彫りにしています。
このブログでは、複数バージョンのDRAGEN Germlineの小規模バリアントコールの精度評価について紹介します。2020年以降、DRAGENメソッドは、precisionFDA Truth Challenge V2およびCMRGに対するバリアントコールのエラーをそれぞれ70%および60%排除しました。これらの結果から、DRAGENは生殖細胞系列バリアントコールとその関連アプリケーションのための非常に説得力のあるソリューションとして位置づけられいます。
WGSとDRAGENの真価を問う
NIST v4.2.1の全ベンチマーク領域の精度
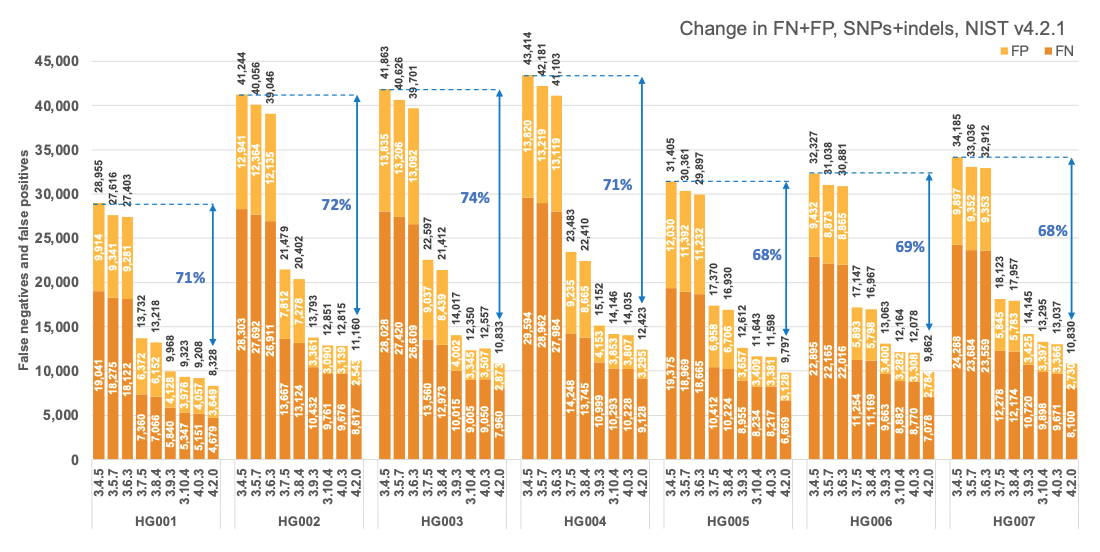
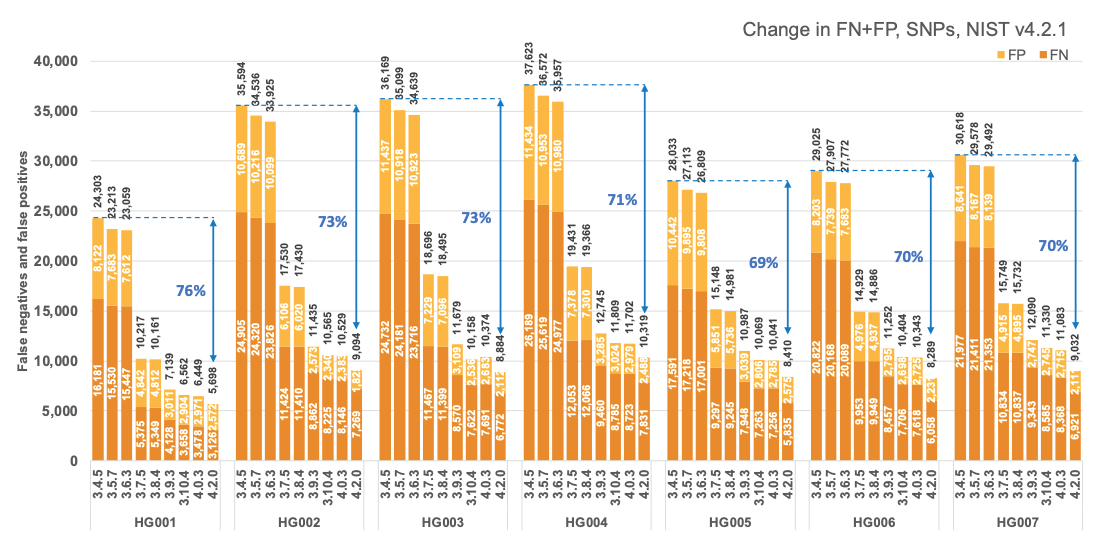
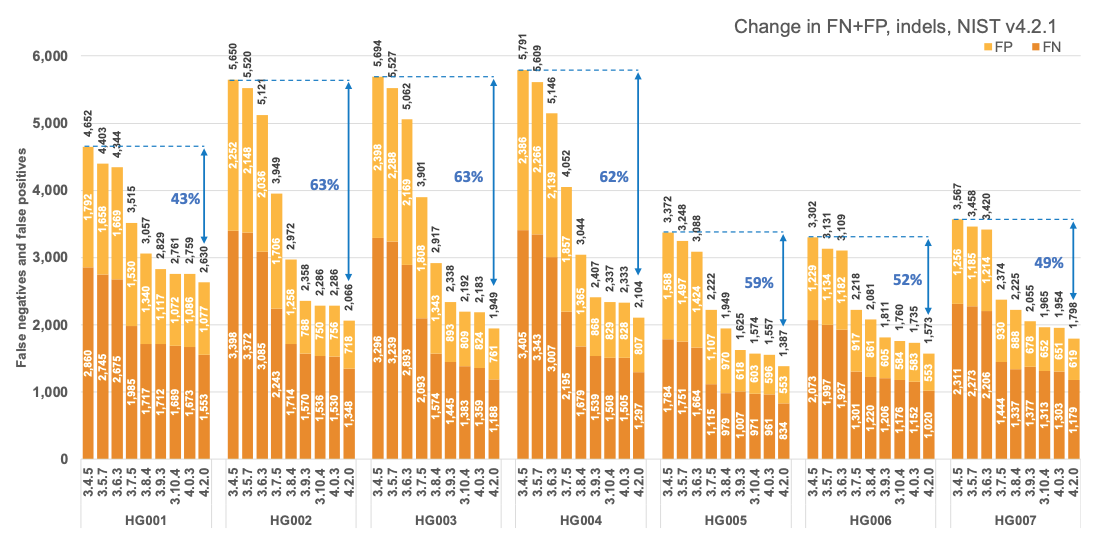
図1は、SNPとIndelの組み合わせについて、v4.2.1 全ベンチマーク領域の7つのNISTサンプル5すべてにおける連続したDRAGENバージョンの正確性を報告しています。DRAGENの精度は時間の経過とともに大幅に改善し、過去2.5年間で平均71%のエラー減少を達成しました。バージョン3.7.5と3.9.3にそれぞれマルチゲノム(グラフ)リファレンスと機械学習(ML)を導入したことで、エラーが急激に減少しました。しかし、さらなる継続的な全体的改善により、DRAGEN v4.2ではv3.4.5と比較して、民族性に関わらず、エラーが約70%減少しました。この改善は7つのNISTサンプルすべてで一貫しています。
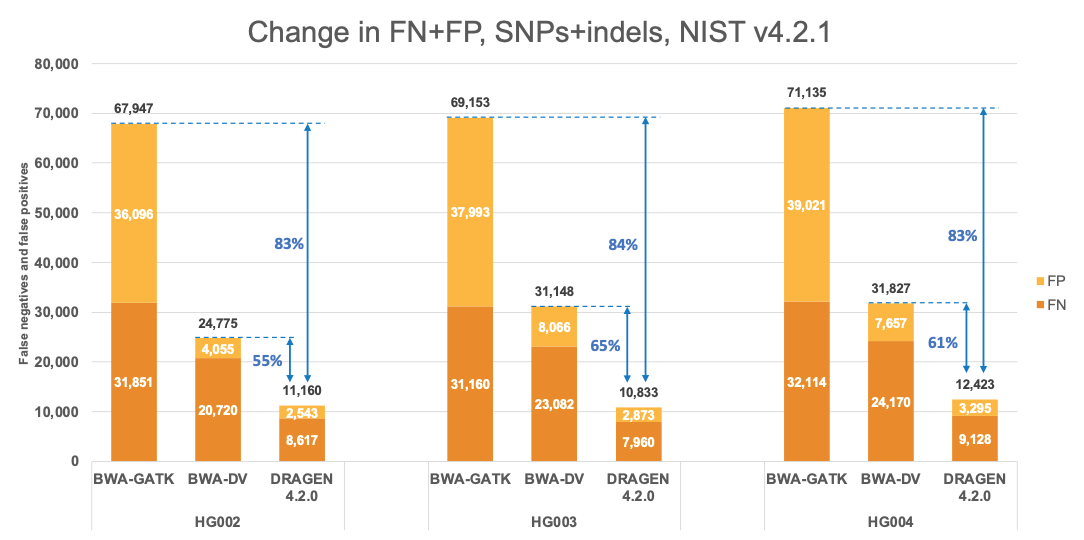
precisionFDA V2チャレンジで選ばれたパイプラインとの精度比較
図4は、v4.2.1 全ベンチマーク領域のprecisionFDA V2サンプル1におけるDRAGEN v4.2の精度を示しています。この精度は、チャレンジ結果9から得られたBWA-GATK6,7およびBWA-DeepVariant6,8 VCFの精度と比較されました。これらは、それぞれに「UYMUW」および「S7K7S」というサブミッションが指定されていました。エラー削減については、DRAGENはSNPとIndelの組み合わせでSNPの平均84%、Indelの平均79%減少という結果が得られ、BWA-GATKを平均83%上回りました。BWA-DeepVariantとの比較では、DRAGENはSNPとIndelの組み合わせで平均60%減少し、SNPの平均62%減少、Indelの平均46%減少という結果でした。これらの結果は公表されたレポートと一致しています10。
Human Pangenome Reference Consortiumによる評価で選ばれたパイプラインとの精度比較
Human Pangenome Reference Consortium(HPRC)は最近、グローバルな遺伝的多様性を表すために選ばれた、47の多様なヒトゲノムの高品質なアセンブルをリリースしました11。これらの47の高品質アセンブルは、単一ゲノムリファレンスによって引き起こされるリファレンスバイアスを低減することを目的としたヒトパンゲノムリファレンスの最初の草案を構成していました。
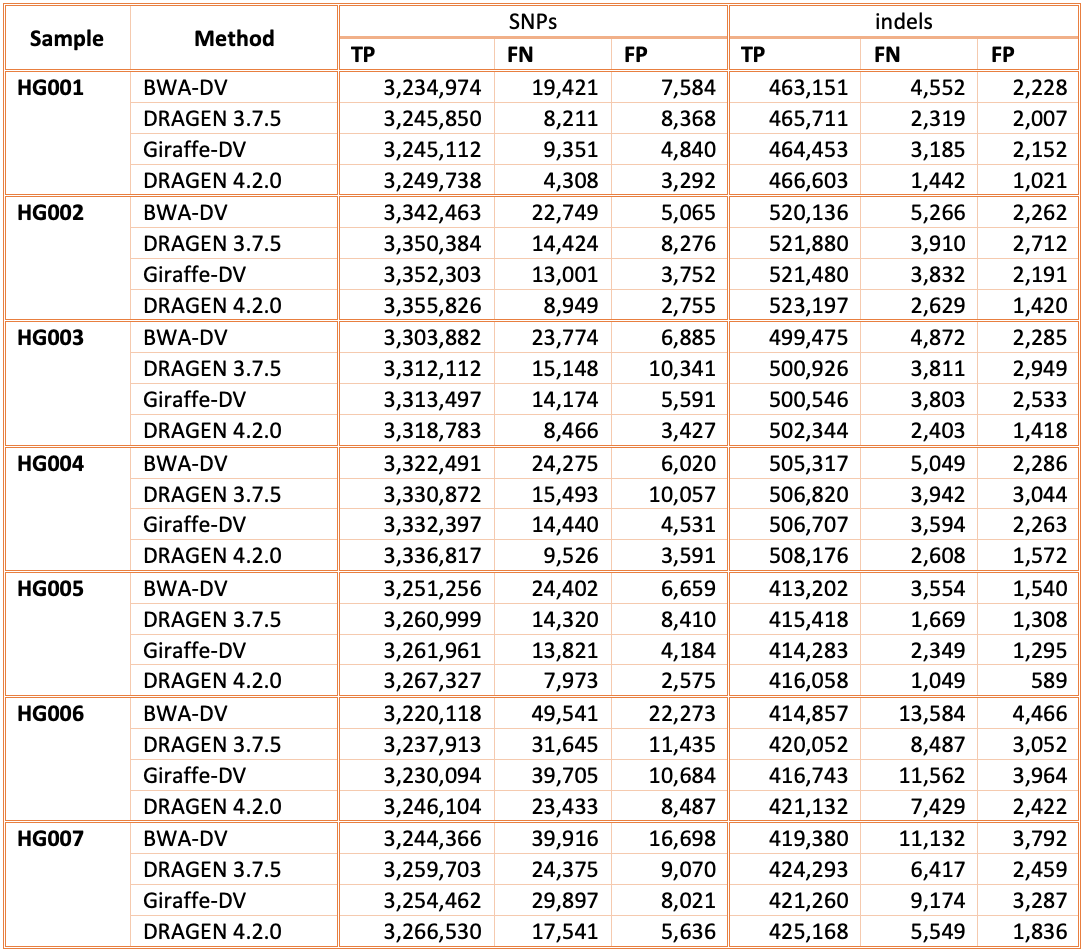
図5と表1では、DRAGEN 4.2の精度を、Giraffe12でHPRCリファレンスパンゲノムにアライメントさせて得られた精度と、v4.2.1 全ベンチマーク領域については、HG001-HG007サンプル上でのDeepVariantおよびBWA-DVパイプラインによるバリアントコールで得られた精度とを比較したものです。Giraffe-DVとBWA-DV VCFの両方はAmazon S3からダウンロードされました13。Giraffe-DVとBWA-DV VCFの生成に使用されたサンプルは30xカバレッジにダウンサンプリングされました。そのため、公正な比較のために、BWA BAMファイルをダウンロードし、DRAGEN 3.7.5とDRAGEN 4.2.0の両方を使用してサンプルを再アライメントしました。BWA-DVと比較すると、DRAGENはSNPとIndelの組み合わせで、SNPでは平均61%、Indelでは平均52%の減少を示し、平均59%のエラー減少を示しました。ここでもprecisionFDA V2サンプルで示された比較で観察された傾向が確認されています。Giraffe-DVと比較した場合、DRAGENはSNPとIndelの組み合わせで、SNPでは平均38%、Indelでは平均41%の減少を示し、平均38%のエラー減少を報告しています。
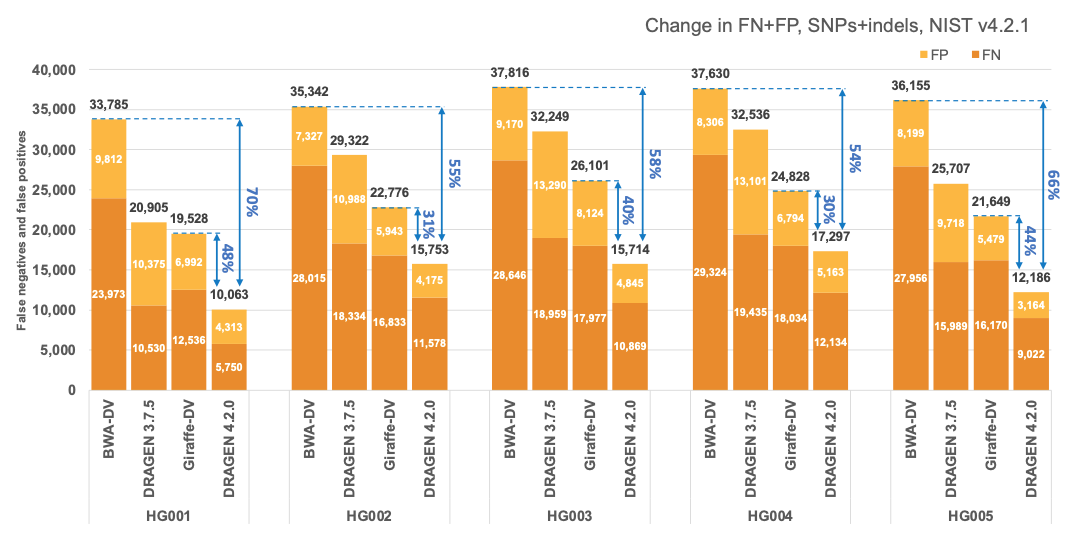
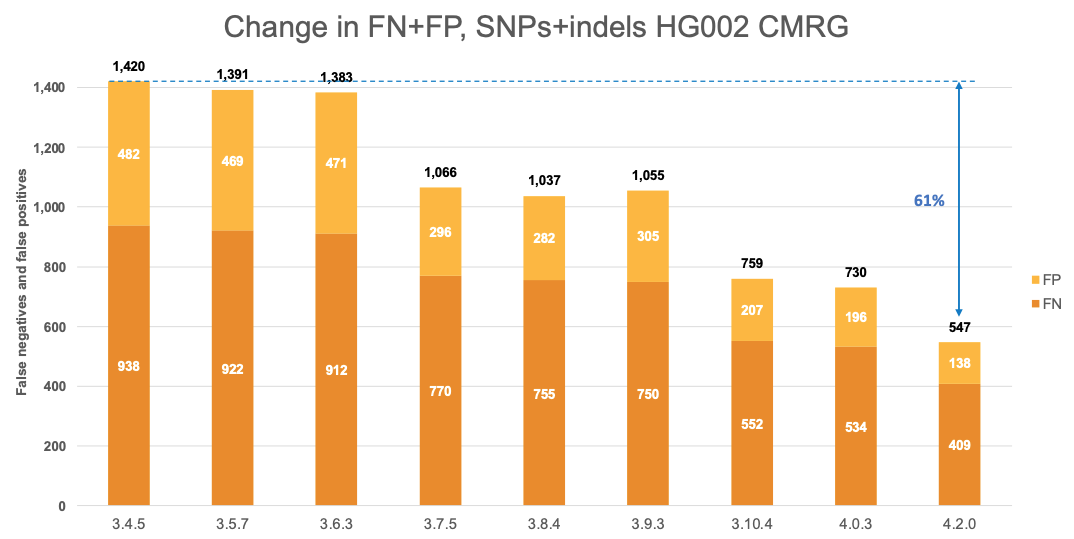
CMRGベンチマーキング領域の精度
図6は、CMRG真理値セット4におけるSNPとIndelの組み合わせを用いたDRAGENバージョンの連続した精度を示しています。NIST v4.2.1 全ベンチマーク領域で観察された精度の向上は、CMRGベンチマーキングセットでも明らかであり、全体的なエラーは61%減少します。この所見は、これらのCMRG領域の遺伝子にみられる反復的または多型的な複雑さにおいても一貫しています。具体的には、バージョン3.7.5ではマルチゲノムリファレンスを導入すると、エラーが23%減少し、その後、バージョン3.10.4ではhg38リファレンスのv2マスキングを行うと、エラーがさらに28%減少しました。バージョン4.2.0の最新のリファレンスおよびマルチゲノム(グラフ)のアップデートにより、エラーがさらに25%減少しました。
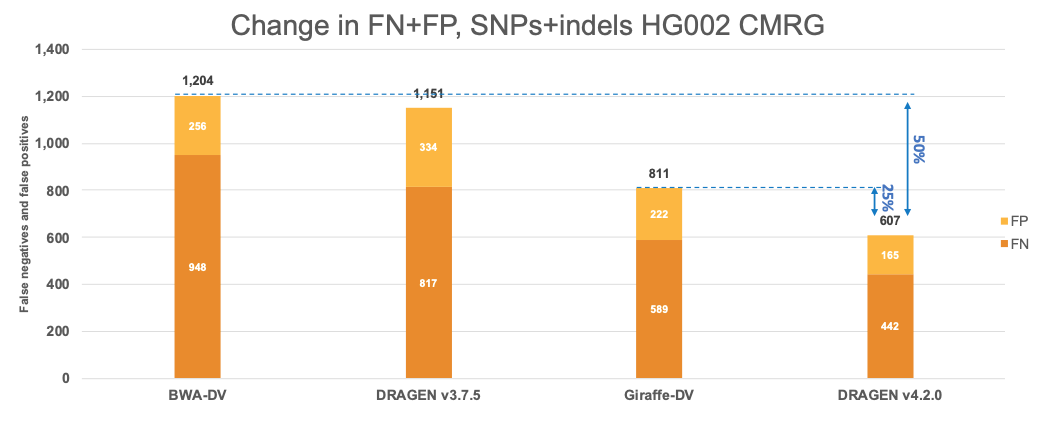
Human Pangenome Reference Consortiumとの比較
図7では、Human Pangenome Reference Consortiumが選定したパイプラインを用いたDRAGENの性能とCMRG真理値セットとの比較に拡大しています。CMRG真理値セットの結果は、全ベンチマーク領域と同じ傾向に従っており、DRAGEN v4.2.0では、BWA-DVと比較して50%、Giraffe-DVと比較して25%の合計エラー減少が示されています。エラー減少は、BWA-DVおよびGiraffe-DVのSNPでそれぞれ55%および25%、Indelでそれぞれ35%および26%に分けられます。
DRAGENバージョン全体にわたる主要機能の進化
2020年以降、DRAGEN Germlineバリアントコールの精度は、マルチゲノム(グラフ)リファレンス(3.7.5)、機械学習の再キャリブレーション(3.9.2、4.0.3で初期設定化)、およびhg38リファレンスの更新などの主要機能の導入により大幅に改善されました14。図8に示すように、これらの主要機能はDRAGENバージョン全体で改良、改善されました。各hg38リファレンスラベルおよびDRAGENマルチゲノム(グラフ)バージョンの定義は、表2のさらに下に記載されています。
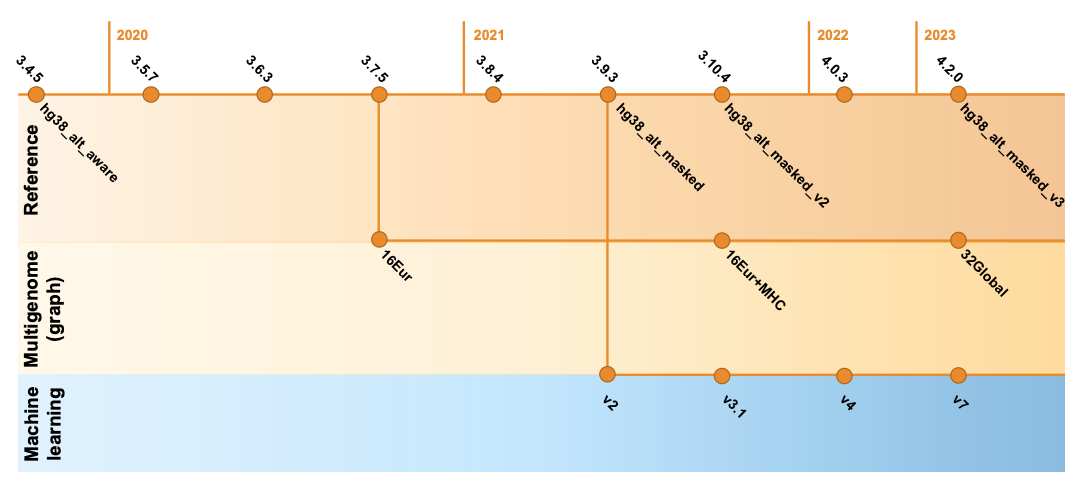
マルチゲノム(グラフ)リファレンス
DRAGENのマルチゲノム(グラフ)リファレンスは、ゲノムマッピングが困難な領域におけるイルミナリードのマッピングと小規模バリアントコーリングの精度を大幅に向上させることが証明されています15,16。
DRAGENマルチゲノム(グラフ)リファレンスでは、標準リファレンスゲノムhg38、hg19、およびhg37d5は、慎重に選択された集団ハプロタイプセグメントで増強され、集団で一般的に発生することが知られている代替経路を介して、高度に多型で相同的領域におけるマッピングの曖昧さを解消するのに役立ちます。これにより、平坦な1次元リファレンスゲノムは、本質的に多くのヒトゲノムのシーケンスによって示された期待に基づいたより複雑な輪郭のマップへと変換されます。例えば、hg38は、マッピング困難領域に800,000を超えるコンティグと400,000を超えるSNPで増強され、10%を超えるゲノムをカバーするようになりました。
ヒト集団全体の遺伝的多様性をより多くキャプチャーするため、DRAGEN v4.2で入手できる最新のマルチゲノム(グラフ)リファレンスは、旧版のヨーロッパ系16集団のサンプルから、世界中の異なる系統の32サンプルに置き換えられました。この新しい「v3」マルチゲノム(グラフ)リファレンスでは、標準リファレンスと比較してゲノムの12%以上が強化され、前バージョンと比較して20%改善されており、民族性バイアスがはるかに少なくなっています。
機械学習の再キャリブレーション
機械学習は、DRAGEN Germlineの小規模バリアントでの精度のもう1つの重要なコンポーネントです15。DRAGEN v4.2では、モデルトレーニングデータを拡張し、エクソームデータを含む深度およびアッセイ構成全体でバリアントコールの精度を向上させました。さらに、リファレンスゲノムオプション全体の性能を強化し、Indelコールを改善し、密集した集積があるためにノイズの多い領域の精度を高める新機能を導入しました。精度の向上だけに満足せず、機械学習(ML)コンピューティングの性能を最適化することができました。アップグレードされたプロセスでは、MLオプションを有効にしても全ゲノムまたはエクソームデータセットのランタイムが著しく増加しない程度まで効率化を実現しました。
GRCh38/hg38リファレンス
hg38リファレンスゲノムは、過去の投稿14およびゲノム生物学17で説明したように、経時的に複数の改訂と改善が行われました。リファレンスゲノムの最新のレビューは、ヒトゲノムの偽陰性のセグメント重複を特定し、未知の領域を明らかにしたchm13の新しい完全なヒトアセンブル18との比較解析によって推進されました。最新のDRAGEN v.4.2では、公開されているGRCh38リファレンスゲノムの最新のアップデートを組み込んだ「hg38-alt-masked-v3」および「hg38-alt-masked-v3-multigenome」の使用を推奨します。
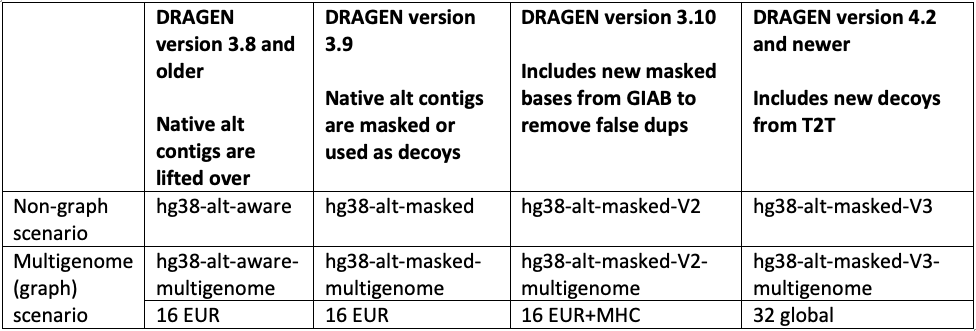
これらすべてのリファレンスのベースラインアセンブルは、European Bioinformatics Instituteからダウンロードされました。19「16 EUR」をヨーロッパ系の16人の個人からのグラフハプロタイプとして、「32 global」をヨーロッパ、アフリカ、アジア、南北アメリカの32人の個人からのグラフハプロタイプとして、および「Native alt contigs」をGRCh38リファレンスゲノムに存在する700+ "*_alt"およびHLAコンティグとして参照しました。
これらの公表されたアップデートに従い、hg38-alt-masked-v3に使用するデコイコンティグとして、chm13およびhs37d5からの別のシーケンスを組み込みました。これらのシーケンスは、重複が崩壊したため4,17、またはシーケンスギャップが不明なため(末端動原体型染色体の短腕のNマスキング)、GRCh38には含めませんでした。これらのデコイコンティグを含めることで、いくつかの技術的に困難な遺伝子(FANCD2、MAP2K3、KCNJ18、KMT2C)やY染色体の領域におけるバリアントコールの精度を向上させることができました。
まとめ
DRAGEN v4.2は、ゲノムの解析が困難な領域を解明するための絶え間ない数々の努力の集大成です。この記事では、リファレンスゲノム、マルチゲノム(グラフ)、機械学習の改善により、最大の課題となっているゲノム領域全体のSNPとIndelコールの精度がどのように大きな飛躍を遂げたのかについて探求しました。これまでの投稿では、ExpansionHunter2のようなクラスベースコーラーやGBA13のような遺伝子の遺伝子特異的コーラーが、その他のタイプの技術的に困難なバリエーションの検索にどのように使用されているのかについて探求しました。
NIST v4.2.1 全ベンチマーク領域とCMRG領域の両方で実施されたテストによると、DRAGEN v4.2は一貫して生殖系列の小規模バリアントを呼び出し、以前のバージョンを上回る優れた精度を示しています。このプラットフォームは、その精確性、速度、スケーラビリティ、遍在性、および包括性で注目を集めており、すべてのゲノミクス解析に不可欠なツールとなっています。DRAGENは、そのユニークな特徴により、ゲノムデータの包括的で効率的な解析を行う研究者やサイエンティストにとって強力なリソースです。
この記事で紹介した結果は、Illuminの高品質シーケンステクノロジーとDRAGENのインフォマティクスを組み合わせることで、技術的に困難な領域を含め、卓越した精度のバリアントコールにつなげる重要点に光を当てています。
DRAGEN v4.2は2023年6月に発売される予定です。
学術用途向けの詳細情報またはDRAGEN試用版ライセンスについては、dragen-info@illumina.comまでお問い合わせください。
DRAGENのコマンドライン
以前のバージョンのDRAGENのDRAGENリソースファイルは、ここからダウンロードできます。テストで使用したコマンドラインオプションを表3と表4に示します。リファレンスハッシュテーブルとMLモデルを表5に示します。
バージョン |
リファレンスハッシュテーブルへのリンク |
MLモデルへのリンク |
3.4.5 |
該当なし |
|
3.5.7 |
該当なし |
|
3.6.3 |
該当なし |
|
3.7.5 |
該当なし |
|
3.8.4 |
該当なし |
|
3.9.3 |
||
3.10.4 |
||
4.0.3 |
V4.0(初期設定) |
|
4.2.0 |
hg38_alt_masked_multigenome_v3 |
V7.0(初期設定) |
表5 該当する各バージョンのリファレンスハッシュテーブルとMLモデルへのリンク。
コマンドラインの例を…で置き換え、リファレンスディレクトリ、出力ディレクトリ、FASTQのサンプルなどのリファレンスラン固有パラメーターを置き換えます:dragen --fastq-file1 ... --fastq-file2 ... --RGSM HG002 –RGID HG002 --ref-dir ... --output-file-prefix HG002 --events-log-file dragen_events.csv --output-directory ... --generate-sa-tags true --enable-vcf-compression true --enable-variant-caller true --enable-map-align true --enable-map-align-output true --enable-sort true --enable-duplicate-marking true --enable-bam-indexing true
NISTの真理値との一致率は、Nature Biotechnology20に記載されているRTGツールキットを使用して実行されます。比較例のコマンドは、次のとおりです。java -Djava.awt.headless=true -Dtalkback=false -Dusage=false -Xmx40g -jar RTG-3.9.1.jar vcfeval -b <truth>.vcf.gz -c <query>.vcf.gz -t <tmp_dir> -o <output_dir>-output-mode annotate -vcf-field QUAL --bed-regions <truth>.bed -Z --sample <truth sample>,<query sample> -ref-overlap
すべてのDRAGENランで、推奨される<sample>.hard-filtered.vcf.gz VCF出力ファイルを使用します。これにより、最良のf1スコア測定値が得られます。
注釈
- N. D. Olson et al., “PrecisionFDA Truth Challenge V2: Calling variants from short and long reads in difficult-to-map regions,” Cell Genomics, vol. 2, no. 5, p. 100129, 2022, doi: https://doi.org/10.1016/j.xgen.2022.100129.
- S. Strom, C.-L. Mead, D. Letchworth, V. Onuchic, and M. Bekritsky, “Fully featured genome: expanding the hunt for genomic variation with DRAGEN STR.” https://www.illumina.com/science/genomics-research/articles/str-expansionhunter.html
- S. Strom, “Using DRAGEN for Gaucher and Parkinson disease research: resolving GBA1 variants using PCR-free whole-genome sequencing.” https://www.illumina.com/science/genomics-research/articles/using-dragen-for-gaucher-and-parkinson-disease-research--resolvi.html
- J. Wagner et al., “Curated variation benchmarks for challenging medically relevant autosomal genes,” Nat Biotechnol, vol. 40, no. 5, pp. 672–680, 2022, doi: 10.1038/s41587-021-01158-1.
- J. Wagner et al., “Benchmarking challenging small variants with linked and long reads,” Cell Genomics, vol. 2, no. 5, p. 100128, May 2022, doi: 10.1016/J.XGEN.2022.100128.
- H. Li, “Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM,” Mar. 2013, Accessed: May 02, 2023. [オンライン] 入手可能: https://arxiv.org/abs/1303.3997v2
- G. A. Van der Auwera and B. D. O’Connor, Genomics in the Cloud : Using Docker, GATK, and WDL in Terra. O’Reilly, 2020.
- R. Poplin et al., “A universal SNP and small-indel variant caller using deep neural networks,” Nature Biotechnology 2018 36:10, vol. 36, no. 10, pp. 983–987, Sep. 2018, doi: 10.1038/nbt.4235.
- “PDR: precisionFDA Truth Challenge V2: Calling variants from short- and long-reads in difficult-to-map regions.” https://data.nist.gov/od/id/mds2-2336 (accessed Apr. 27, 2023).
- S. Zhao, O. Agafonov, A. Azab, T. Stokowy, and E. Hovig, “Accuracy and efficiency of germline variant calling pipelines for human genome data,” Scientific Reports 2020 10:1, vol. 10, no. 1, pp. 1–12, Nov. 2020, doi: 10.1038/s41598-020-77218-4.
- W.-W. Liao et al., “A draft human pangenome reference,” Nature 2023 617:7960, vol. 617, no. 7960, pp. 312–324, May 2023, doi: 10.1038/s41586-023-05896-x.
- J. Sirén et al., “Pangenomics enables genotyping of known structural variants in 5202 diverse genomes,” Science, vol. 374, no. 6574, pp. 1-11, Dec. 2021, doi: 10.1126/SCIENCE.ABG8871.
- “A draft human pangenome reference – Variant calls” s3://human-pangenomics/publications/PANGENOME_2022/DeepVariant/samples (accessed May 18, 2023).
- S. Catreux et al., “Demystifying the versions of GRCh38/hg38 Reference Genomes, how they are used in DRAGEN and their impact on accuracy.” https://www.illumina.com/science/genomics-research/articles/dragen-demystifying-reference-genomes.html
- S. Catreux et al., “DRAGEN Sets New Standard for Data Accuracy in PrecisionFDA Benchmark Data.” https://www.illumina.com/science/genomics-research/articles/dragen-shines-again-precisionfda-truth-challenge-v2.html
- R. Mehio et al., “DRAGEN Wins at PrecisionFDA Truth Challenge V2 Showcase Accuracy Gains From Alt-aware Mapping and Graph Reference Genomes.” https://www.illumina.com/science/genomics-research/articles/dragen-wins-precisionfda-challenge-accuracy-gains.html
- S. Behera et al., “FixItFelix: improving genomic analysis by fixing reference errors,” Genome Biol, vol. 24, no. 1, p. 31, 2023, doi: 10.1186/s13059-023-02863-7.
- S. Nurk et al., “The complete sequence of a human genome,” Science (1979), vol. 376, no. 6588, pp. 44–53, Apr. 2022, doi: 10.1126/SCIENCE.ABJ6987.
- “GRCh38 Full Analysis Set Plus Decoys HLA.” http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/GRCh38_reference_genome/GRCh38_full_analysis_set_plus_decoy_hla.fa (accessed Mar. 30, 2023).
- P. Krusche et al., “Best practices for benchmarking germline small-variant calls in human genomes,” Nature Biotechnology 2019 37:5, vol. 37, no. 5, pp. 555–560, Mar. 2019, doi: 10.1038/s41587-019-0054-x.