はじめに
次世代シーケンサー(NGS)テクノロジーは、全ゲノムシーケンス(WGS)と完全なバリアント情報に容易にアクセスできるようにすることで、研究者が集団遺伝学研究を行う方法を変革しました。しかし、強力な全ゲノム関連解析(GWAS)に必要な数百から数千のサンプルから高いカバレッジのシーケンスデータを生成すると、コストが大幅に上昇し、その可能性が制限されます。最近、低カバレッジWGSに続けてインピュテーションを行うアプローチが、一般的かつ低頻度なバリエーションをキャプチャーするための費用対効果が高く、有用なアプローチとして登場しました1-3。これは特に標準的な一塩基多型(SNP)アレイが十分に表現されていない集団において特に有用です4。
遺伝型インピュテーションは、リファレンスハプロタイプパネルからの情報を使用してデータを精緻化し、シーケンス深度の低いデータセットにおける間隔の広いリード間のギャップを埋めます。この方法により、研究者は大量の個人サンプルでジェノタイピング未実施のバリアントを推測することができ、GWASの統計的検出力を向上できる可能性があります。ゲノム予測の重要なステップは、インピュテーションの精度であり、これは下流解析に大きな影響を与える可能性があります。インピュテーションの精度に影響を与える主な要因には、リファレンスパネルのサイズと位置カバレッジ、インピュテーションの方法、インピュテーション対象のバリアントのマイナーアリル頻度、ハプロタイプフェージングの精度などがあります5。
Illumina DRAGEN™ Bio-IT Platform上で、「遺伝型尤度インピューテーションフェージング法」(GLIMPSE)6のインピュテーション精度とソフトウェアアクセラレーションを組み合わせ、低カバレッジシーケンスデータを補完して両アリルSNPバリアントコーリング性能を最適化しました(図1)。アクセラレーテッドGLIMPSEインピュテーションツールをDRAGENプラットフォームに統合すると、計算時間が40%短縮され、複数の染色体に対して単一のコマンドを使用するエンドツーエンドなパイプラインでスケーラビリティが得られます。さらに、当社のインピュテーションワークフローでは、イルミナのIRPv1リファレンスパネルを活用して、バリアントコーリングの性能をさらに高めています。この記事では、GLIMPSEのDRAGEN実装が偽のコールの総数をどのように減少させるかについて論じ、インピュテーションテーブルおよび入力サンプル数がインピュテーション精度をどのように向上させるかを示すデータを提示します。
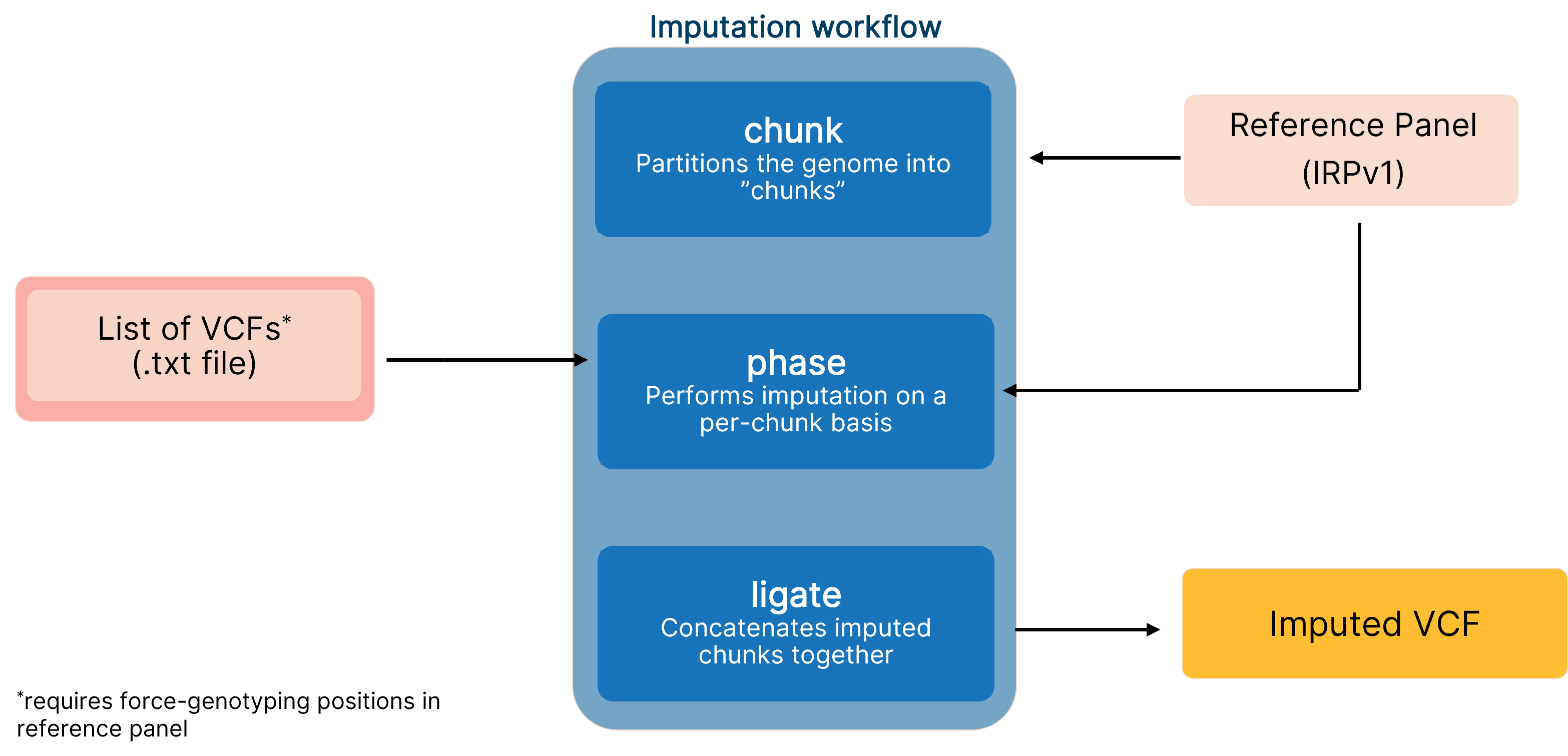
DRAGENへのGLIMPSEの実装を使用して、低カバレッジシーケンスデータを補完するエンドツーエンドなパイプラインです。これは、IRPv1リファレンスパネルを活用してバリアントコール性能を向上します。
DRAGENへのGLIMPSEの実装により、補完されたカバレッジ1×データにおけるバリアントコーリング性能が向上
使用するインピュテーション手法またはアルゴリズムは、インピュテーションデータの品質に直接影響を与えます7。 GLIMPSEはオープンソースのインピュテーションソフトウェアツールで、リファレンスパネルのすべての変異座位においてGibbsサンプラースキームを使用して高品質のコンセンサスベースのハプロタイプコーリングを生成します6。このツールの解析タイムラインをさらに加速し、単一のコマンドライン命令を使用してインピュテーション効率を向上させるために、Illumina DRAGEN v4.0に高精度のGLIMPSEを導入しました。DRAGEN v4.0のソフトウェア高速化コードにより、GLIMPSE Phase v1.1.1と比較して、インピュテーションランタイムが40%短縮しました(図2)。
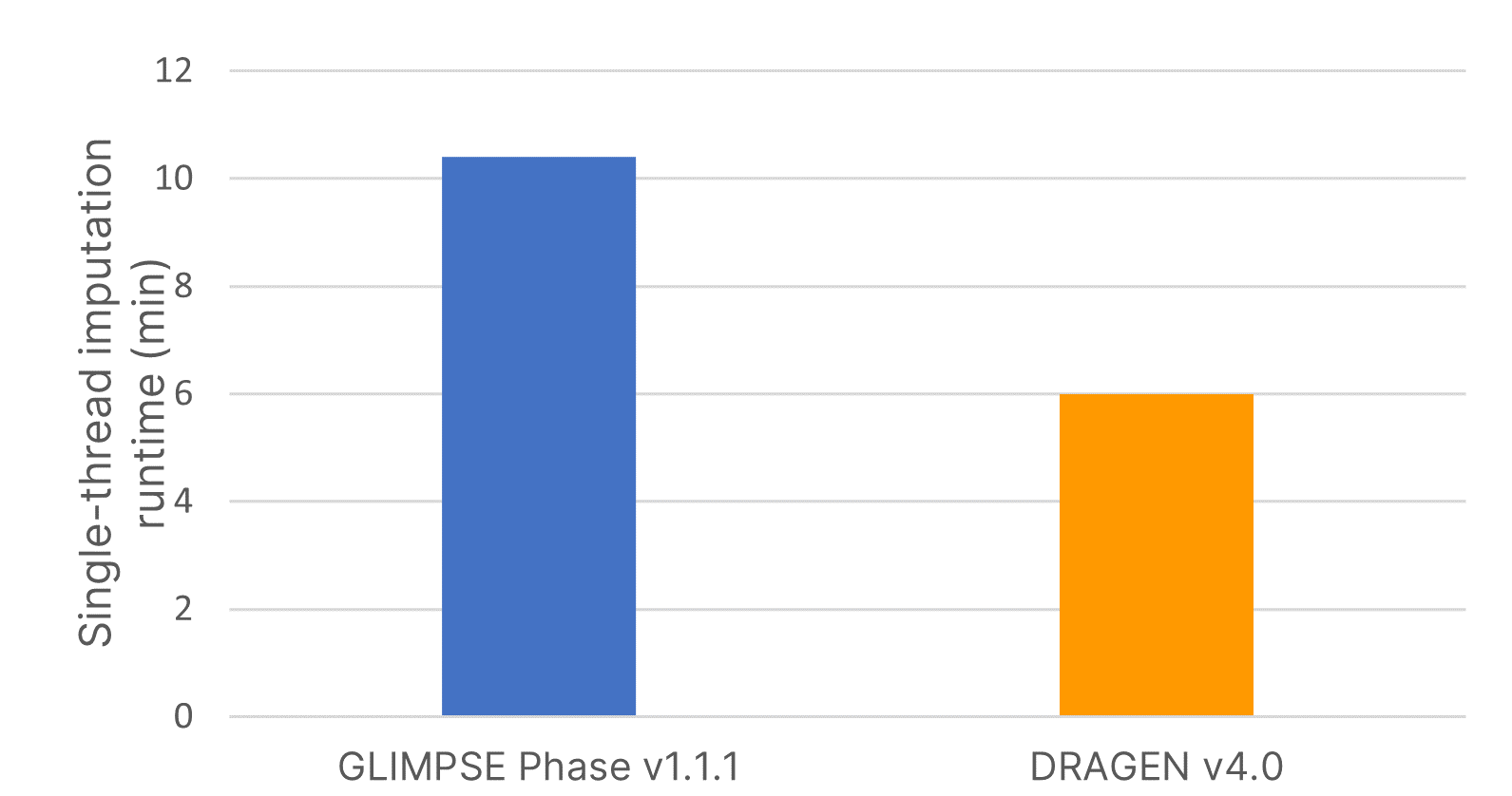
2489サンプルからなる大規模かつ多様なIRPv1リファレンスパネルを使用し、83,172座位を含む領域のインピュテーションを行うために、15サンプルのインプットバリアントコール形式(VCF)ファイルを使用してインピュテーションのランタイムを評価しました。ソフトウェア高速化インピュテーションにより、GLIMPSE Phase v1.1.1と比較して計算時間が1.7倍速くなりました。
一般的なインピュテーション性能を評価するために、Genome-in-a-Bottle(GIAB)コンソーシアム8,9の3つのサンプルHG002、HG003、HG004を使用し、1×カバレッジにダウンサンプリングしました。これらのサンプルは、シーケンスパイプラインを検証し、新しいバリアントコーリング法を開発するために広く使用されている、十分に特性評価されたヒトゲノムです。真理値データセットはNCBIベンチマークSNP(v4.2.1)で構成されていました8,9。
この小さなサンプルセットにおいて、インピュテーションなしの低カバレッジ(1×)データとインピュテーション後のデータの総偽コール率を比較した結果、IRPv1リファレンスパネルを使用してインピュテーションを行った各サンプルにおいて偽コール率が約90%減少していることが明らかになりました。これは主に偽陰性(FN)コールの減少によるものです(図3)。当社の調査結果は、DRAGEN v4.0に導入されたGLIMPSEが、非常に効率的な遺伝子型インピュテーションとバリアントコール性能の向上を提供することを実証しています。
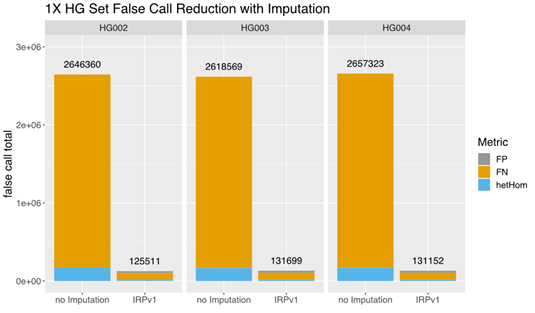
DRAGEN v4.0に導入されたGLIMPSEツールとIRPv1リファレンスパネルを使用して、インピュテーションを行った場合と行わなかった場合の、低カバレッジ(1×)シーケンスデータ間の総偽コール率を比較しました。このアプローチを用いたインピュテーションにより、評価した各サンプル(HG002、HG003、HG004)で偽コール率が約90%低下しました。FP:偽陽性、FN:偽陰性、hetHom:ヘテロ接合/ホモ接合比。
IRPv1リファレンスパネルがインピュテーション精度を向上
インピュテーションは、ハプロタイプ共有の原理に基づいており、信頼度の高い遺伝子型情報を持つトレーニングセットを使用して、欠損した遺伝子を予測します。その結果、どのインピュテーションツールの精度も、ジェノタイピング未実施のバリアントを推測するために使用されるリファレンスパネルの集団構造に大きく依存します7,10。新しいリファレンスパネルIRPv1を開発し、GLIMPSEによるバリアントコール性能に対するインピュテーションリファレンスパネルのサイズと遺伝的多様性の影響を実証しました。
IRPv1は、1000 Genomes Projectからの2,489サンプルを含む常染色体SNPリファレンスパネルで、小児サンプルを除外するようにトリミングされています。これらの高度にベンチマークされたサンプルは、DRAGEN v3.7.6を使用して、GRCh38ヒトリファレンスゲノムに対して約50×カバレッジのニューヨークゲノムセンターのデータからバリアントコールしたものです。シングルトンSNPおよびHardy-Weinberg平衡から外れていると観察されたSNPを除外しました。複数の代替アリルが観察されたSNPについては、最も頻繁に観察された代替アリルのみを保持しました。SNPは、SHAPEIT411を用いてフェージングしました。
バリアントコール性能に対するリファレンスパネル属性の影響を評価するために、以下の3つのリファレンスパネルをテストしました。
· IRPv1:26集団グループからの2,489サンプルで構成される大規模パネル
· IRPv1-100:IRPv1からの100サンプルのサブセットで構成される小規模、高密度パネル
· IRPv1-PJL:パキスタンのラホールにあるパンジャビ集団(PJL)からの96サンプルを含む遺伝学的に層別化された小規模リファレンスパネル
アフリカ人(AFR)、東アジア人(EAS)、ヨーロッパ人(EUR)の3つの上位集団から5つのサンプルを選び、1×カバレッジでテストを実施しました。真理値データセットは、同じサンプルをDRAGENで50×カバレッジ処理したデータを基準として定義しました(表1)。偏りのない解析を保証するため、インピュテーションに使用されたこれらの15サンプルはIRPv1リファレンスパネルから除外しました。IRPv1-100の100サンプルのサブセットは、IPRv1の集団グループ間の多様性がこの小規模パネルで維持されるように選択されました。

AFR:アフリカ人、EAS:東アジア人、EUR:ヨーロッパ人。
各パネルに表示される位置の総数は以下のとおりです。
· IRPv1:49,493,544
· IRPv1-100:22,066,395
· IRPv1-PJL:13,429,942
当社の結果(図4)によると、完全なIRPv1リファレンスパネルではインピュテーション性能が最も高かったのに対して、縮小版パネルでは偽陽性率の増加とともに感度が低下したことが示されました。この性能の低下はAFR集団で最も顕著であり、集団レベルではハプロタイプ構造により多くの不均一性が見られました7,12。このテスト集団では、AFR集団と遺伝的に異なるIRPv1-PJLリファレンスパネルをインピュテーションに用いた場合、感度が最も低く、偽陽性率が最も高い結果となりました。これらの結果は、大規模で遺伝的に多様なリファレンスパネルを使用して、観察されたハプロタイプがリファレンスデータセットに存在する可能性を最大化し、全体的なインピュテーション精度を向上させることの重要性を強調しています。
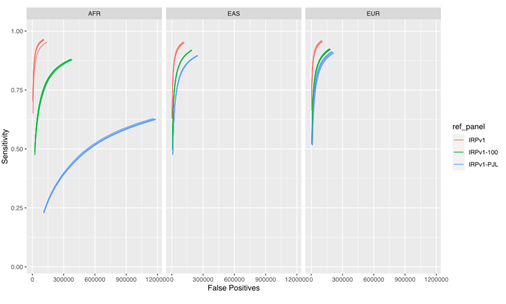
3つの上位集団(AFR、EAS、EUR)について、3つのリファレンスパネル(IRPv1、IRPv1-100、IRPv1-PJL)を用いて感度と偽陽性バリアントコール率を計算しました。全体的なインピュテーション精度とバリアントコーリング性能は、欠損している遺伝型の推定に使用されるリファレンスパネルのサイズと遺伝的多様性に依存していました。AFR:アフリカ人、SAS:東アジア人、EUR:ヨーロッパ人の集団
マルチサンプルモードによりインピュテーション性能が向上
GLIMPSEツールにより、研究者は単一サンプルまたはマルチサンプルモードで解析を実行し、最適ではないリファレンスパネルでも、シーケンス深度の低い(1×カバレッジ)入力データを使用してバリアントコーリング性能を向上させることができます。マルチサンプルモードでは、ソフトウェアへのインプットとしてマルチサンプルVCFファイルが提供されます。インピュテーションツールはサンプルを一度に1つずつインピュテーションしますが、マルチサンプルVCF内のその他のサンプルをリファレンスパネルに組み込み、これらのサンプルを追加のリファレンスデータポイントとして利用することで、ハプロタイプブロックの定義とマッチングを支援します。研究者がインプットとして単一サンプルVCFファイルのリストを提供するオプションを追加しました。このリストは、マルチサンプルVCFファイルが提供されたかのようにマルチサンプルモードで実行されるため、マルチサンプルモードで処理可能な入力の範囲が拡張されます。
サンプルを個別に実行する場合とマルチサンプルモードで実行する場合の影響を評価するために、前述のAFR上位集団からの5つのサンプルを対象に、3種類のリファレンスパネル(IRPv1、IRPv1-100、IRPv1-PJL)を使用する場合と、遺伝的に層別化されたIRPv1-PJLリファレンスパネルのみを使用する場合のテストを実施しました。真理値データセットは、50×カバレッジの同じサンプルで構成されていました。より大規模で多様性の高いリファレンスパネルを使用した場合、サンプルモードがインピュテーション精度に与える影響は最小限でした。しかし、本研究の結果から、遺伝的多様性が低いリファレンスパネルであっても、低カバレッジサンプルにマルチサンプルモードを適用することで感度が向上し、インピュテーション精度が改善されることが示されました(図5)。
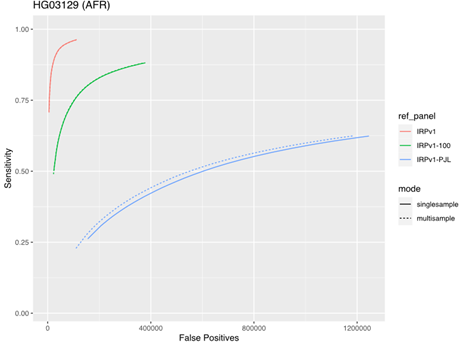
3つのリファレンスパネル(IRPv1、IRPv1-100、IRPv1-PJL)を用いて、遺伝的に多様なアフリカ人(AFR)上位集団からの5つのサンプルについて、感度と偽陽性バリアントコール率を計算しました。インピュテーションは、単一サンプルまたはマルチサンプルモードで実施されました。全体的なインピュテーション精度とバリアントコーリング性能は、最適ではないリファレンスパネルを使用しても、多様なAFR集団に対してマルチサンプルモードでサンプルを実行することで向上しましたが、多様なパネルでサンプルを実行する場合、最高の性能が得られました。
まとめ
この記事では、大規模な低カバレッジデータセットからのジェノタイピングを補完するために使用されるGLIMPSEツールの主な機能強化について説明しました。まず、高精度なGLIMPSEソフトウェアスイートがDRAGEN v4.0に組み込まれ、解析速度が向上しており、単一のコマンドライン命令で実行できます。当社の修正ワークフローを使用したインピュテーションは、低カバレッジデータをインピュテーションする場合に、バリアントコーリング性能を劇的に向上させます。しかし、全体的なインピュテーション精度は、観察されていない遺伝型を推測するための適切なリファレンスパネルに依存しています。当社は、26のサンプルグループからなる2489サンプルを含む、大規模で遺伝的多様性の高いリファレンスパネルIRPv1を開発しました。これを高精度なインピュテーションツールと組み合わせることで、3つの上位集団においてテストされたバリアントコーリング性能をさらに向上させました。特に、リファレンスパネルが小さく、テスト集団に存在するすべてのハプロタイプをキャプチャーするために十分な多様性がない場合に、マルチサンプルモードを使用すると、さらにインピュテーション精度が向上します。
学術用途向けの詳細情報またはDRAGEN試用版ライセンスについては、dragen-info@illumina.comまでお問い合わせください。
注釈
1. Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11(7):499-511. doi:10.1038/nrg2796
2. Pasaniuc B, Rohland N, McLaren PJ, et al. Extremely low-coverage sequencing and imputation increases power for genome-wide association studies. Nat Genet. 2012;44(6):631-635. doi:10.1038/ng.2283
3. Naj AC. Genotype Imputation in Genome-Wide Association Studies. Curr Protoc Hum Genet. 2019;102(1):e84. doi:10.1002/cphg.84
4. Martin AR, Atkinson EG, Chapman SB, et al. Low-coverage sequencing cost-effectively detects known and novel variation in underrepresented populations. Am J Hum Genet. 2021;108(4):656-668. doi:10.1016/j.ajhg.2021.03.012
5. Zhang B, Zhi D, Zhang K, Gao G, Limdi NN, Liu N. Practical Consideration of Genotype Imputation: Sample Size, Window Size, Reference Choice, and Untyped Rate. Stat Interface. 2011;4(3):339-352. doi: 10.4310/sii.2011.v4.n3.a8.
6. Rubinacci S, Ribeiro DM, Hofmeister RJ, Delaneau O. Efficient phasing and imputation of low-coverage sequencing data using large reference panels. Nat Genet. 2021;53(1):120-126. doi:10.1038/s41588-020-00756-0
7. Schurz H, Müller SJ, van Helden PD, et al. Evaluating the Accuracy of Imputation Methods in a Five-Way Admixed Population. Front Genet. 2019;10:34. doi:10.3389/fgene.2019.00034
8. Zook JM, Catoe D, McDaniel J, et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci Data. 2016;3:160025. doi:10.1038/sdata.2016.25.
9. Wagner J, Olson ND, Harris L, et al. Benchmarking challenging small variants with linked and long reads. Cell Genomics. 2022;2(5):100128. doi:10.1016/j.xgen.2022.100128.
10. Ahmad M, Sinha A, Ghosh S, et al. Inclusion of Population-specific Reference Panel from India to the 1000 Genomes Phase 3 Panel Improves Imputation Accuracy. Sci Rep. 2017;7(1):6733. doi:10.1038/s41598-017-06905-6
11. Delaneau O, Zagury JF, Robinson MR, Marchini JL, Dermitzakis ET. Accurate, scalable and integrative haplotype estimation. Nat Commun. 2019;10(1):5436. doi:10.1038/s41467-019-13225-y
12. Xu ZM, Rüeger S, Zwyer M, et al. Using population-specific add-on polymorphisms to improve genotype imputation in underrepresented populations. PLoS Comput Biol. 2022;18(1):e1009628. doi:10.1371/journal.pcbi.1009628