はじめに
次世代シーケンサー(NGS)により、サイエンティストはゲノムを解読し、生物学をより深く理解することができます。実績のあるイルミナのsequencing by synthesis(SBS)ケミストリーと受賞歴のあるDRAGEN二次解析を組み合わせることで、全ゲノムシーケンス(WGS)データを卓越した精度で実現します。1,2 DRAGEN Multigenome(グラフ)は、困難な領域におけるマッピング精度を約50%もさらに向上させます。1それでも、ショートリードだけではマッピングが難しい、より長いリード長のマッピング可能性の向上から利益を得ることができる少数のジェニック領域が残っています。
イルミナの完全ロングリードは、ロングリードシーケンスをアクセシブルにし、ヒトゲノムのこれらの困難な領域を解決するための合理化されたワークフローを提供します。イルミナの完全ロングリードを使用すると、1つのプラットフォームからショートリードとロングリードが可能です。DRAGENインフォマティクスと機械学習手法を組み合わせることで、Illumina Complete Long ReadsはNGSテクノロジーから正確なバリアントコーリングとフェージング情報を抽出します。この記事では、Illumina Complete Long Readヒトゲノム解析の背後にある基本原則を掘り下げています。
仕組み:アッセイの概要
Illumina Complete Long Readsワークフロー(図1)は、独自のライブラリー調製アッセイ、実績のあるIllumina SBSケミストリー、強力なDRAGEN二次解析を組み合わせ、5~7 kbのN50で高精度のロングリードデータを生成します。
イルミナ完全ロングリードのライブラリー調製
効率的な1日ライブラリー調製プロトコールは、ハイスループット研究のための拡張が容易で、50 ngのDNAインプットのみを必要とします。* このアッセイでは、タグメンテーションを使用して長いgenomic DNA断片(> 10 kb)を作製し、追加のせん断やサイズ選択の必要性を排除します。長い単一分子DNA断片は、単一塩基対変化の固有のパターンで酵素的にマークされます。これらのランドマークは、DNA断片の長さに沿って低頻度(4%~7%)で導入されます。各単一分子断片には、(複雑なバーコードやアダプターを使用せずに)長いリード情報を取得して保存するためのランドマークの固有の署名があります。ランドマーク長鎖断片を増幅し、続いて2番目のタグメンテーションステップを行い、イルミナシステム上で標準シーケンス用のライブラリーを調製します。
* 50 ngのDNAインプットが推奨されます。10 ngのDNAインプットが可能です。
バイオインフォマティクスワークフロー
解析パイプラインは、長いリードを生成し、データを標準の未マークWGSライブラリー†と組み合わせて、元の単一分子断片の完全かつ正確な表現である長い連続したリードを生成します。
† 解析には、同じサンプルから30倍の標準ショートリードヒト全ゲノムデータが必要です。Illumina DNA PCR-Free Prepを推奨します。サードパーティ製WGSキットにも互換性があります。マークされていないライブラリーは、並行して調製またはシーケンスする必要はありません。以前にランしたサンプルからFASTQファイルを使用することができます。
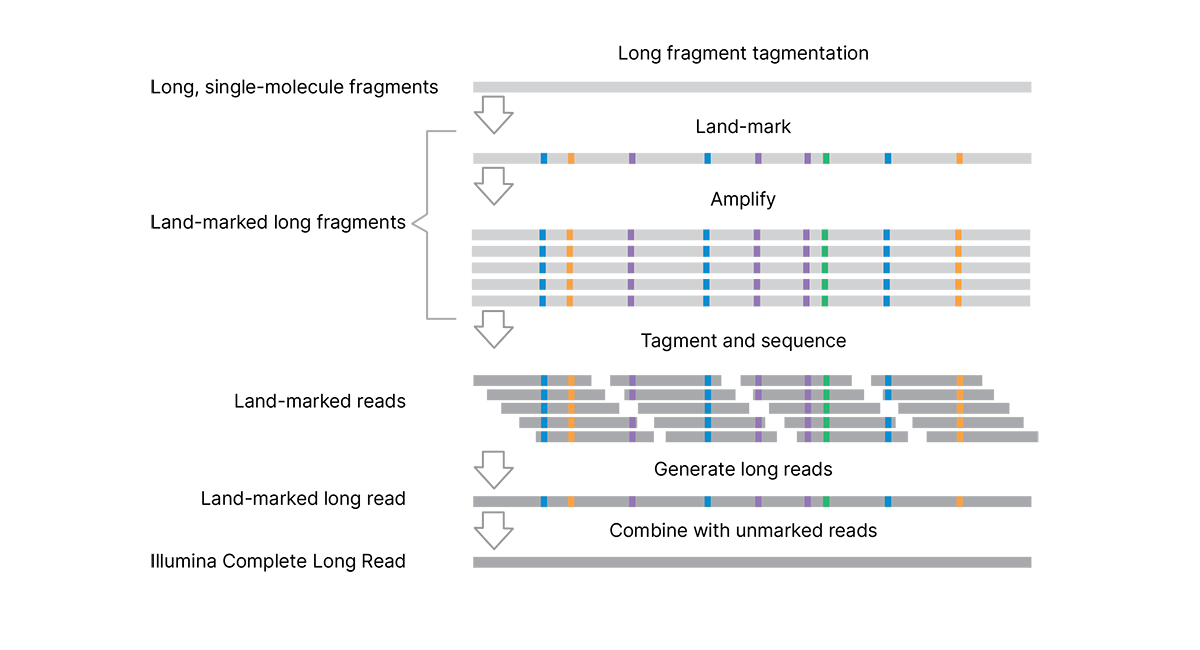
このアッセイでは、タグメンテーションを使用して長いDNA断片を作製し、せん断やサイズ選択の必要性を排除します。長鎖断片は、一分子スケールでランドマーク化されており、断片内の長鎖情報を取得して保持します。ランドマーク長鎖断片を増幅し、続いて2番目のタグメンテーションステップを行い、シーケンス用のライブラリーを調製します。解析パイプラインはロングリードを生成し、データを標準的でマークなしのWGSライブラリー(同じサンプルから、別個にシーケンス)と組み合わせて、ランドマークを除去し、高精度の完全なロングリードを生成します。
> 仕組みを見る
イルミナの完全ロングリード世代
イルミナのロングリードバイオインフォマティクスのロングリード生成ワークフローには、アライメントやバリアントコーリングなどの標準的なゲノム計算手法が含まれます。ワークフローは、BaseSpace Sequence Hubでプッシュボタンアプリとしてパッケージ化され、利用できます。このワークフローでは、ランドマーク付きライブラリーと非マーカーライブラリー、およびリファレンスゲノムを入力として使用します。これらのインプットは、一連のステップ(図2)を実行するために使用され、包括的なWGS解析のために単一分子からロングリードを生成します。

リード上のランドマークサイトの特定
ロングリード生成プロセスの最初のステップは、ランドマークライブラリーに存在するマークを特定することです。マップに自信のある領域では、ほとんどのランドマークは標準アライメントとリファレンスゲノムとは異なるヌクレオチドの検出によって同定できます。
リファレンスゲノムに容易にアラインできない領域(反復領域など)からのリードでは、ランドマークを検出するために異なるアプローチが必要です。k-merの特定の方法(すなわち、k長のヌクレオチドの小さなストリングにリードを非公式に分割する)により、アルゴリズムはリファレンスゲノムを使用せずにリード間の関係を決定できます。マップが困難な領域では、マークは、ランドマークリードとマークなしリードのk-merを比較することで推測されます。3マークありリードのk-merが、マークなしリードのk-merとペアにできない場合、それはランドマークとして扱われます。
ランドマーク化されたリードの加重ネットワークの構築
リード内のすべてのランドマークを検出した後、次のステップは、リード間の接続を共有マークに基づいて特定することです。当社では、類似するリードのペアのインデックスを作成し、k-merマッチングを最適化するために、minimator k-merを使用しています。4 特定のminimator k-merを共有するすべてのペアを詳細に比較できます。共有および競合するランドマークの数によって、リードを結びつけるエビデンスの強度が決まります(図3)。当社は、これらの接続の強度に基づいて、マークされたリードの加重ネットワークを構築します。
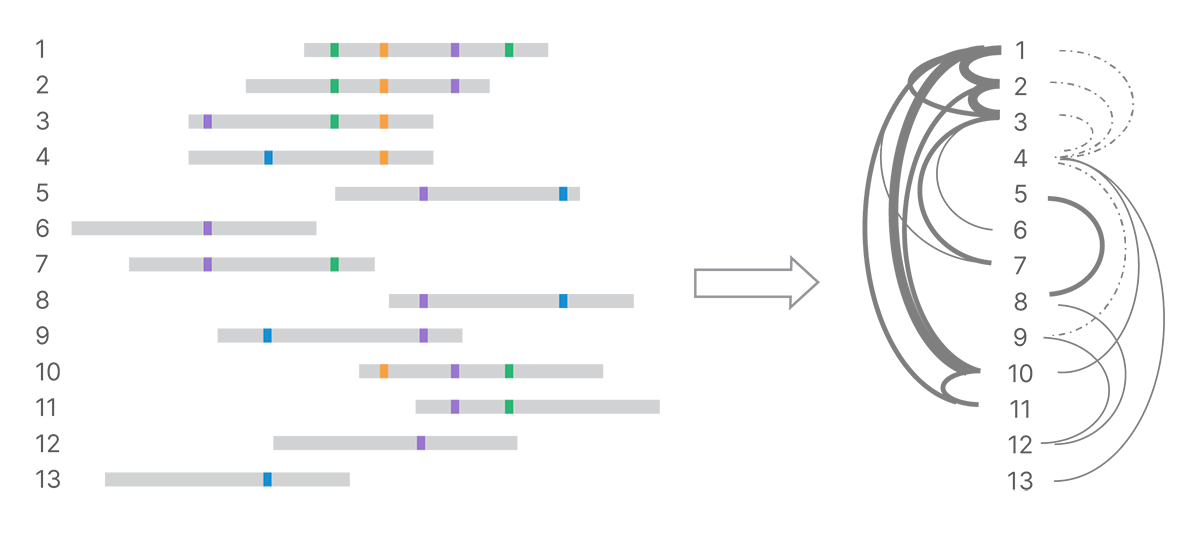
共有のランドマークはリードをネットワークに接続し、共有のランドマークの数と競合するランドマークの数に応じて接続の強さが異なります。右側のグラフでは、より強い接続は太い線で示され、より弱い接続は点線で示されています。
同じテンプレートからリードのグループを見つける
リード間の接続は、すべてのリードのグラフを形成します。一連の分解およびクラスター化手法(共有されたランドマーク数が少ないために競合する接続や弱い接続を除去するなど)を適用して、ネットワーク全体を強くリンクされたクラスターに分割します(図4)。各クラスターは単一分子に由来すると推定されます。
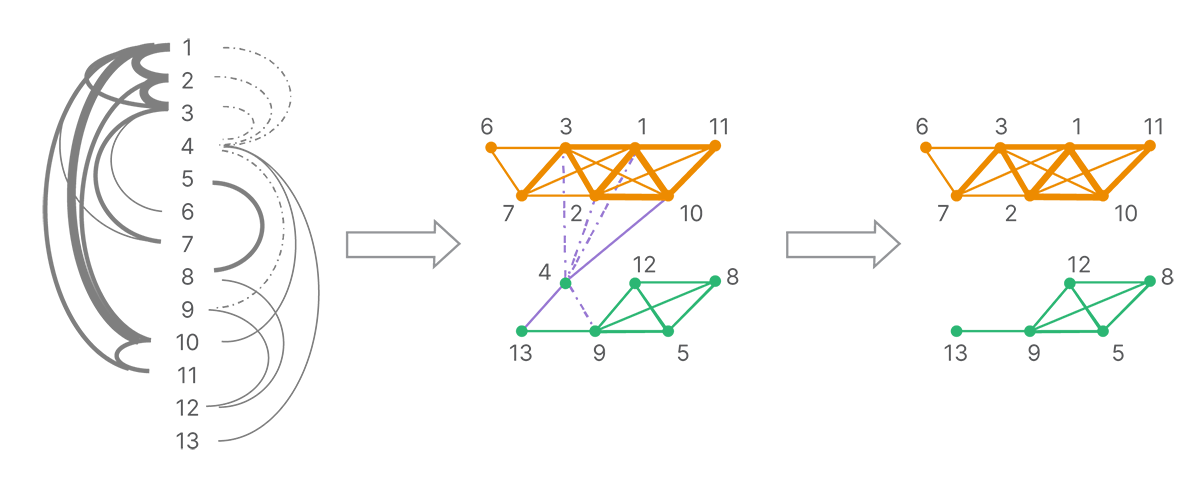
接続のグラフは、最も強い接続に従って分割されます。強く接続された各クラスターは、単一のテンプレート分子からのリードで構成されると推定されます。
各リードのグループをアセンブルします
最終的なクラスターから、DRAGEN解析ではk-merベースのde Bruijnグラフのようなアセンブリ手法を使用して、ロングリードコンティグを生成します(図5)。
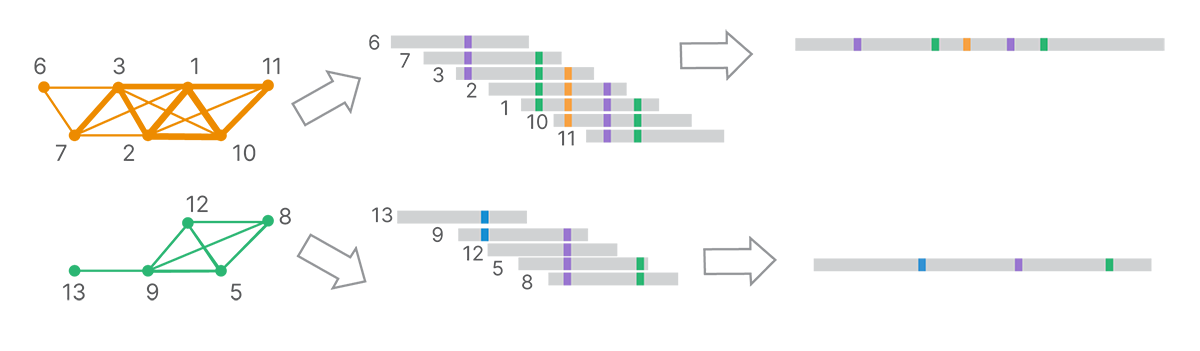
1つのテンプレート分子から得られると推測されるリードのセットに対応する各クラスターは、ランドマークされたロングリードに組み立てられます。
ロングリードからランドマークを削除
長いリードの生成をサポートするためにランドマークを使用した後、マークを取り除くことができます。ランドマークと真のバリアントを区別するために、ランドマークロングリードを非マーカーリードと比較します。対応する未マーキングリードと一致しないランドマークは、最終のIllumina Complete Long Readが真のシーケンスを明らかにするように更新されます(図6)。ランドマーク付きロングリードとノンマークリードの比較は、特にマッピングが困難な地域では、部分的にリファレンスゲノムアライメントを使用し、部分的にk-merインデックスを使用して、ランドマークの同定方法に似ています。ノーマークリードとランドマークロングリードのアライメントを取得した後、ベイズモデルを適用してロングリードの最終ベースコールと対応するクオリティスコアを決定します。
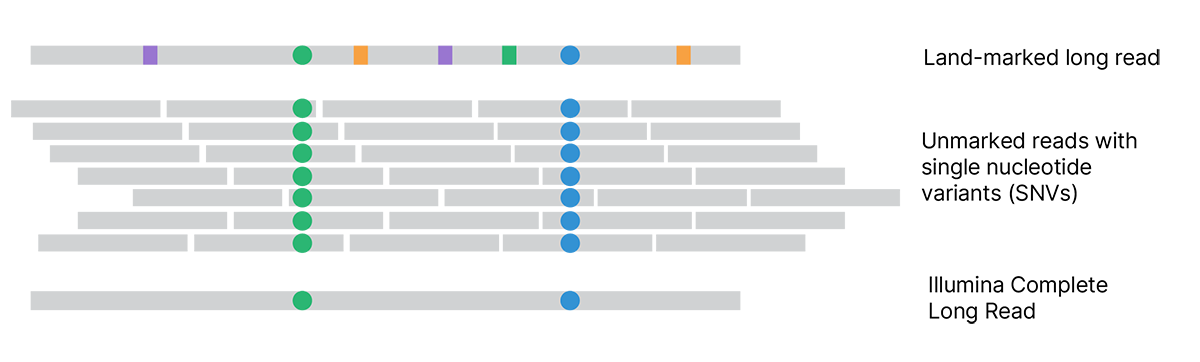
各アセンブルされたランドマーク付きロングリードをノンマークリードと比較し、ランドマーク(四角)と真のバリアント(円)を区別します。マークなしリードと競合するランドマーク塩基は、マークなしリードと一致するように更新されます。最終的なイルミナの完全ロングリードは、元の単一分子を正確に表し、真のシーケンスを明らかにします。
二次解析
上記のIllumina Complete Long Read構築ステップの後、イルミナの完全ロングリードとマークされていないショートリードが二次解析に使用されます(図7)。完全ロングリードは、まずMinimap2の改変バージョンを使用してゲノムにアライメントされます。
小さなバリアントコーリングの場合、ロングリードとショートリードのDRAGENの小さなバリアントコーリングの結果は、単一のVCFファイルにマージされます。DRAGEN Small variant callは、75 kbを超えるリードを処理できます。機械学習モデル(Genome in a Bottleからのバリアントコールでトレーニング)は、ロングリードとスタンダードショートリードから得られた小さなバリアントコールを組み合わせて改善するために使用されます。最後に、WhatsHapの修正版は、イルミナの完全ロングリードのフェージングに使用され、小さなバリアントをハプロタイプ情報をキャプチャするために作成された新しい包括的な出力ファイルとマージしました。
構造バリアントコーリングの場合、ロングリード構造バリアントコーラー(Sniffles2)出力5およびショートリードDRAGEN構造バリアントコーラーの結果が単一のVCFファイルにマージされます。
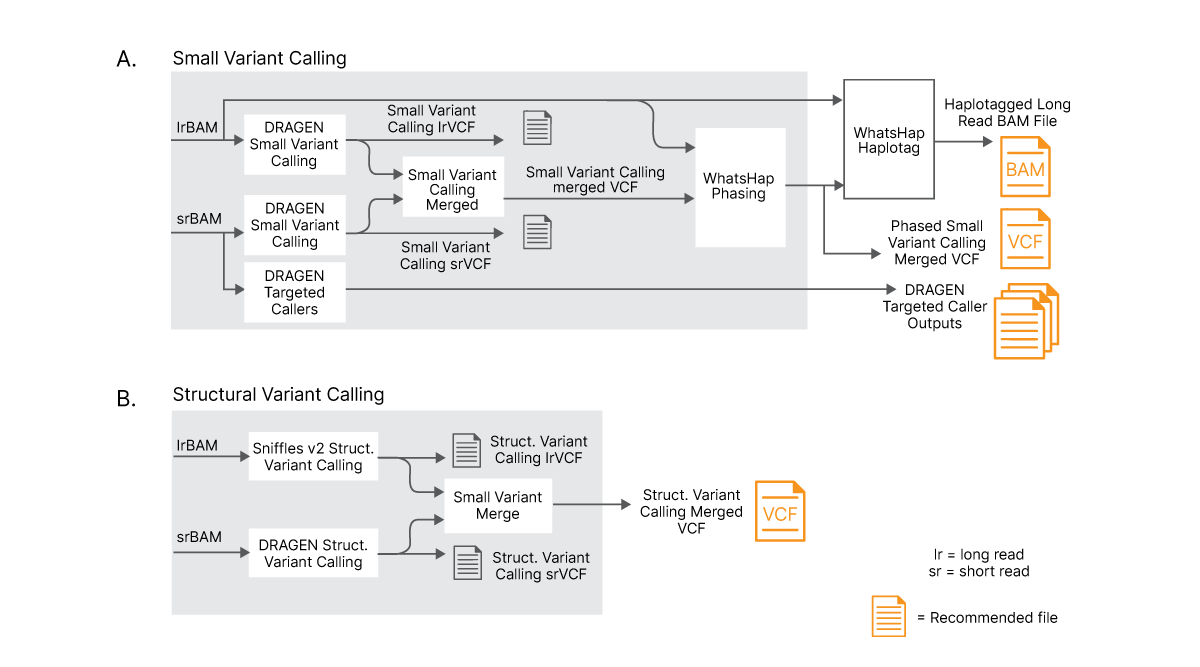
(A)ロングリードとショートリードは別々にアライメントされ、結果は高度なロジックと組み合わされてバリアントコーリングを最適化します。長いリードとマージされた小さなバリアントは、フェージングツールを使用してフェーズされます。(B)ロングリードとショートリードは、専用のSVコーラーで構造バリアント(SV)コールを実行するために別個に使用され、結果は新しい統合SV VCFファイルを作成するために高度なロジックを使用して統合されます。
高精度WGS
Illumina Complete Long Readテクノロジーは、実績のあるIllumina SBSケミストリーとDRAGEN二次解析を活用して、ヒトWGSの精度をさらに向上させます。PrecisionFDA Truth Challenge v2データセットでは、Illumina Complete Long Readアッセイを使用したWGSの精度と想起を反映したF1スコアは99.87%でした(図8)。6,7 標準的なWGSと比較して、Illumina Complete Long Readデータは、複数のベンチマークサンプルにわたるSNPとインデルの両方における偽陰性と偽陽性の全体的な減少を示しています(図9)。
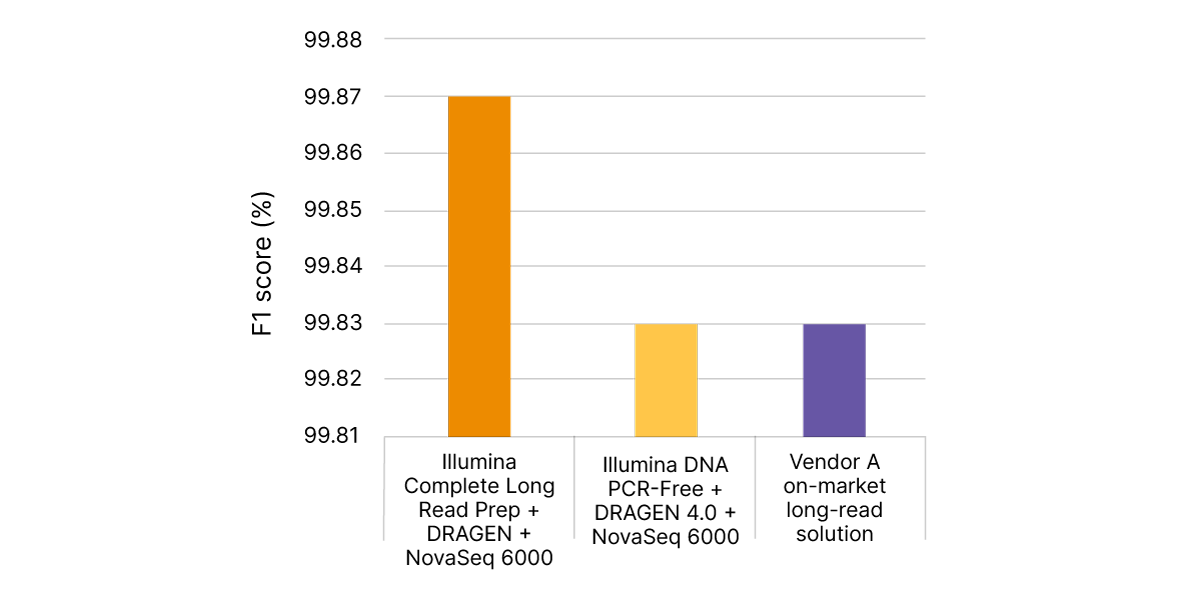
PrecisionFDA Truth Challenge v2データセットにより、Illumina Complete Long Read Prep, Human(オレンジ)はF1スコア(%)で測定した高精度のバリアントコーリングを提供し、WGSの精度とリコールを反映しています。Illumina DNA PCR-Free PrepおよびDRAGEN 4.0(黄色)または市販の別のロングリードソリューション(紫色)を使用した標準WGSは、この精度と一致しません。
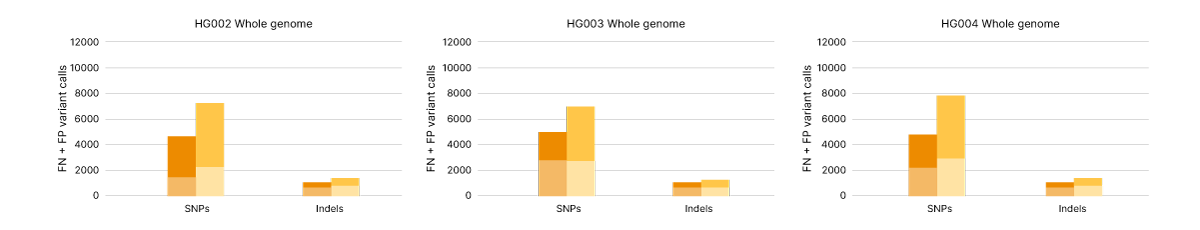
ボトルヒトリファレンスサンプルHG002, HG003HG004のゲノムの偽陽性(FP)および偽陰性(FN)として測定される一塩基多型(SNP)およびIndelバリアントコーリング精度。イルIllumina Complete Long Readアッセイ(オレンジ)とIllumina DNA PCR-Free Prep(黄色)のWGSデータをゲノム全体で比較する。
結論
ロングリード情報は、ゲノムの最も困難な領域を解決するのに役立ちます。イルミナの完全ロングリードは、同じ装置でロングリードとショートリードの両方を可能にすることで、ゲノミクスラボが包括的なWGSに簡単にアクセスできるようにします。イルミナのコンプリートロングリードは、効率的で使い慣れたラボワークフロー、最小限のインプット要件、大規模ライブラリーキット製造、遺伝子領域全体で高品質で包括的なバリアントコールを生成するための連続リードなどの利点を提供します。
詳細はこちら
Illumina Complete Long Read製品ライン
イルミナの完全ロングリードの使用が、いかに小さなバリアントコーリングの精度を向上させるかをご覧ください:イルIllumina Complete Long Read調製による包括的WGS、ヒューマンテクニカルノート
参考文献
- Mehio R, Ruehle M, Catreux S, et al. DRAGENがPrecision- FDA Truth Challenge V2で勝利。Alt-Aware MappingとGraph Reference Genomeからの精度向上が明らかになりました。 2023年5月16日にアクセス。
- Illumina. DRAGEN Bio-IT Platformによる生殖細胞系列の小さなバリアントコールの精度向上。 2023年5月16日にアクセス。
- Leinonen M, Salmela L. 間隔を空けたシードを使用した長いk-merの抽出。 IEEE/ACM Trans Compu Biol Bioinform。2022;19(6):3444-3455。Doi:10.1109/TCBB.2021.3113131
- Roberts M, Hayes W, Hunt BR, Mount SM, Yorke JA. 生物学的シーケンス比較のための保管要件の削減。 Bioinformatics. 2004;20(18):3363-3369. doi:10.1093/bioinformatics/bth408
- Sedlazeck FJ, Rescheneder P, Smolka M, et al. 単一分子シーケンスによる複雑な構造変化の正確な検出。 Nat Methods. 2018;15(6):461-468. doi:10.1038/s41592-018-0001-7
- Illumina. 社内資料。2022年。
- PrecisionFDA. Truth Challenge V2: Calling Variants from Short and Long Reads in Difficult-to-Map Regions. precision.fda.gov/ challenges/10. 2023年1月12日にアクセス。