はじめに
がんゲノミクスアプリケーションでは、全ゲノムシーケンス(WGS)を使用して、腫瘍サンプルの完全な変異プロファイルを検出できます。同じ個体からの正常組織サンプルが利用できる場合、体細胞がんバリアントは、正常に存在しない腫瘍サンプルから抽出されたDNAに存在するものとして特定できます。しかし、がんサンプルの不均一な性質やサンプルの純度が低いなどの交絡の影響により、体細胞バリアントはさまざまな頻度で存在するため、正確なバリアントコールが困難になります。
低頻度のバリアントは、サポートが少数のシーケンスリードに限定され、低レベルのエラーはバリアントの解釈ミスとなり、偽陽性のコールにつながる可能性があるため、自信を持って同定することは特に困難です。理想的には、偽陽性コールを最小限に抑え、治癒の負担を軽減し、正確な患者の診断と治療選択を推進したいと考えています。これは、バリアントコーリングアルゴリズムのストリンジェンシーを高めることで達成できますが、これは真のバリアント(偽陰性)の欠損にもつながります。体細胞バリアントコーラーは、偽陽性と偽陰性の間の最良のトレードオフをゲノム全体で実現するように最適化されていますが、ゲノムの一部は、他のものよりも偽コールを生成することが知られています。平均的なパフォーマンスを最大化するためにバリアントコーラーをチューニングすると、特定の地域ではアンダーパフォームにつながります。
我々は、サンプルコホートにおいて、系統的に高品質または低品質のゲノム領域を経験的に定義する方法1を開発しました。これにより、サンプルのコホートからパフォーマンスメトリクスを集約することで、システマティックシーケンスのパフォーマンスを追跡することができます。この解析では、これらのパフォーマンスメトリクスには塩基品質、マッピング品質、平均シーケンス深度が含まれます。これらのメトリクスを使用して、各ゲノム位置における通常のシーケンス品質からの偏差の意義をしっかりと定量化することができます。その後、閾値を導き出し、ゲノムの各位置を高品質または低品質としてラベル付けし、これらのラベルを使用してバリアントコール性能を層別化することができます。
ここでは、がんバリアントコールにこの手法を適用し、偽陽性と偽陰性の密度が地域の系統的な品質変動によってどのように影響を受けるかを示します。
結果
1. アレル頻度による体細胞一塩基多型(SNP)の検出限界の モデリング
その最もシンプルな形態では、小さなバリアントの呼び出し元の目的は、各サイトで非リファレンスベースコールがノイズか、またはアリルからのシグナルかを識別することです。体細胞バリアントコーラーは倍数性を仮定しないため、予想されるバリアントアリル頻度(VAF)に制約はありません。発呼者が特定の周波数でバリアントを正しく同定する能力は、サンプリング要因によって制限されており、シグナルとエラーを累積分布関数(CDF)による二項分布としてモデル化することができます。
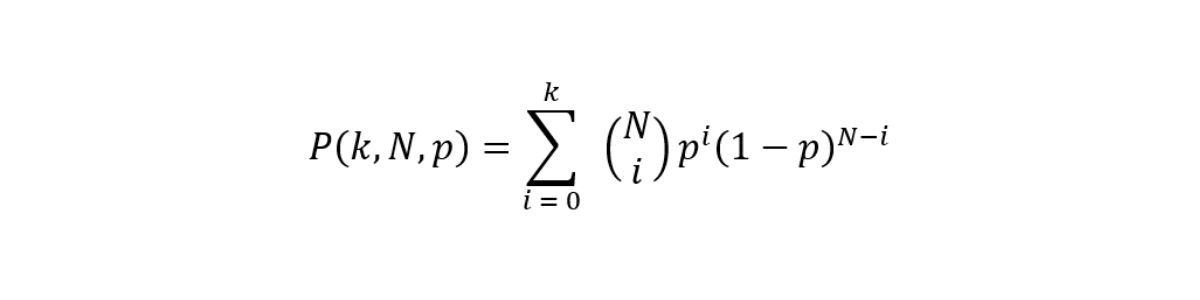
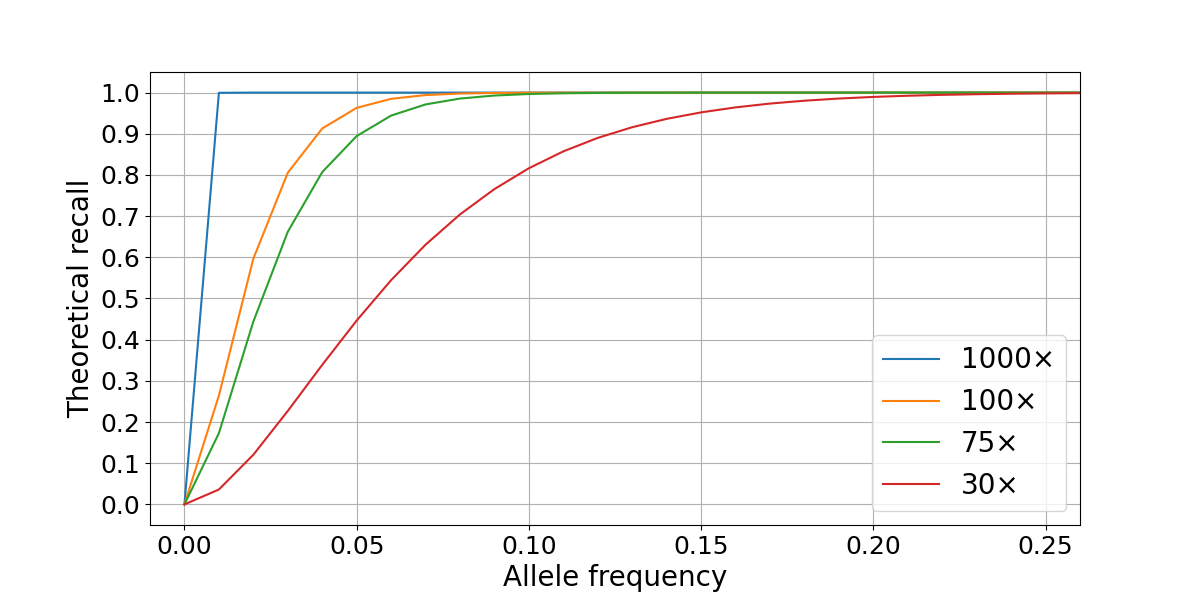
図1 :理論的想起は、Q30の固定値に最も近い閾値を使用して、シグナルの生存関数として計算されます。4本の線は異なるレベルのシーケンス深度(30×、75×、100×、1000×)を表します。
このシナリオでは、サンプルを30×でシーケンスするだけで、≥ 0.2のVAFを持つバリアントを確実に検出できますが、0.1 VAF以下のバリアントを呼び出すには、より深いシーケンスが必要です。実際、シーケンスコストはカバレッジ拡大の限界要因であり、腫瘍シーケンス深度は100倍と75倍で、性能とコストの間で最高の妥協点となります。バリアントコーリングの精度は、サンプリング要因だけでなく、ゲノムにおけるローカルカバレッジの変動や系統的バイアスの影響も受けます。このモデルを使用して、ゲノムの高品質領域と低品質領域における当社の性能を比較します。
2. 混合サンプルによるアレル頻度の シミュレーション
さまざまなVAFにおけるWGSの体細胞リコールを評価するために、Platinum Genomesプロジェクト2の一環として作成された精選された真実のセットを活用しました。これらは、メンデルハプロタイプの一貫性を使用して導き出された家系の13人のメンバーのための信頼性の高い生殖細胞系列コールセットで構成されています。家系の2人の親メンバー(NA12877とNA12878)のシーケンスデータを異なる割合でマージすることで、異なる純度レベルでシミュレーションした腫瘍サンプルを生成しました3。これにより、シミュレートされたバリアントアリル頻度の範囲がもたらされます。例えば、40% NA12878~60% NA12878の混合では、腫瘍として使用されるサンプルからのヘテロ接合バリアント(NA12878)が(完全)混合生物学的サンプルに0.2の予想VAFで存在し、ホモ接合バリアントは0.4のVAFで存在しています。私たちの実験では、2つのシーケンスデータセット間のサンプリングの違いなどの要因により、真の周波数がこれらの理論値から逸脱する可能性があります。腫瘍サンプル(NA12878)にプライベートなバリアントのみを含み、予想されるVAFでアノテーションされた、各純度レベルについて一連の混合真相セットを生成しました。DRAGEN Somatic Pipeline v3.6.3 4を使用して、シミュレーションされた腫瘍(混合サンプル)に対する体細胞コールを作製しました。真のバリアントの想起は、rtgtools vcfevalパッケージ5を使用して設定された真実と比較して、VAFの範囲とシーケンス深度で計算されました。混合データセットでは、SNPリコールはVAF≦0.1では有意に低下するが、この損失はシーケンス深度が高いほど部分的に軽減されることがわかりました(図2)。
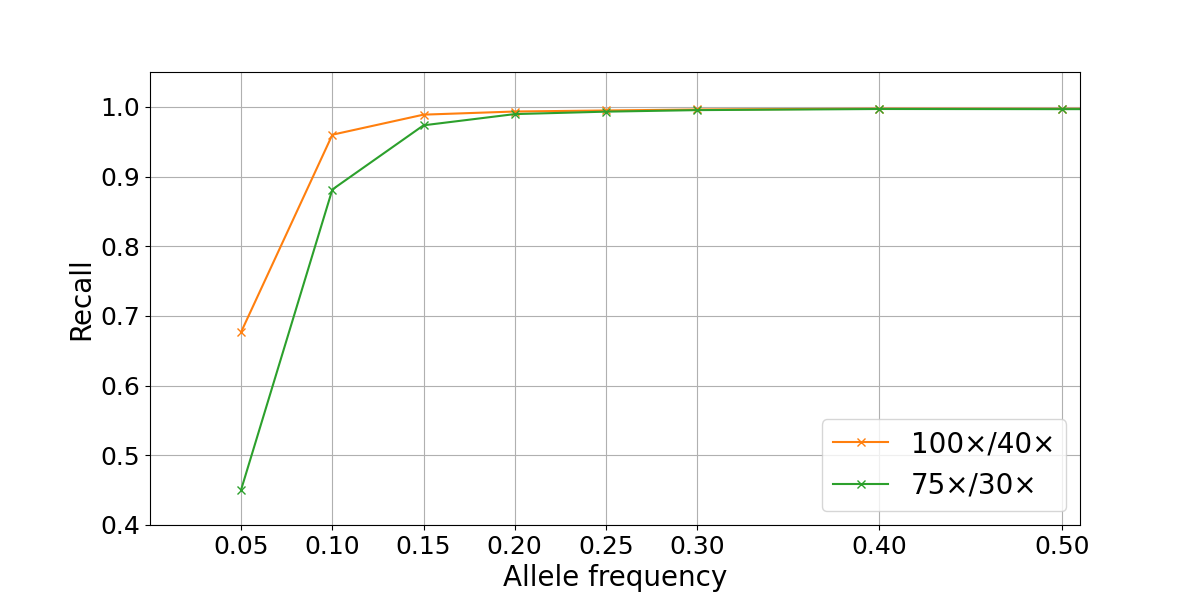
図2:NA12878/NA12877混合物におけるVAFによる 体細胞SNPのリコール。リコールは、2つのシーケンス深度の組み合わせ(シミュレーションされた腫瘍サンプルと正常サンプルでは100×/40×および75×/30×)についてのみ、常染色体上で計算されます。
3. 系統的品質によるゲノムのセグメント化
過去のブログ投稿1で提案したように、多くのWGSサンプルに集約された塩基ごとのシーケンス品質メトリクスを使用して、異常なゲノム領域を特定することができます。具体的には、各メトリクスについて、代表的なゲノムワイド分布を構築し、ベイズガウス混合モデルをそれに当てはめ、非標準的挙動を持つ部位を分布の外れ値として特定しました。その後、ゲノムの各位置を高品質または低品質としてラベル付けしました。マッピングの質、塩基コーリングの質、深度が標準的な挙動を示したゲノム位置は、高品質(N以外の常染色体の89.9% - 2,477 Mb)とラベル付けされ、3つのメトリクスのうち少なくとも1つがゲノム全体の分布から有意な偏差を示した位置は、低品質(N以外の常染色体の10.1% - 279 Mb)とラベル付けされました(表1)。解析は、ヒトゲノムのN以外の常染色体領域に限定されました。
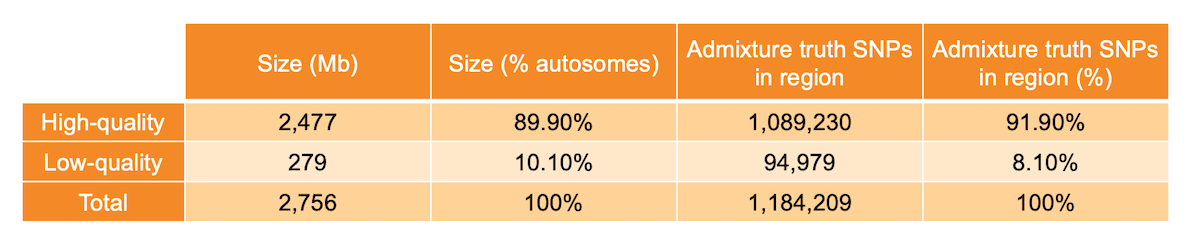
表1: 高品質および低品質の領域のサイズと、調査対象領域に存在する混合SNPの数。
4. 地域の品質による体細胞回収の層別化
2つの異なる深度構成で以前に定義された高品質領域と低品質領域におけるSNPリコールを調べました。試験した両方の深度レベルについて、高品質の領域で計算したリコールは、上記の理論上のリコールを約0.15のアリル頻度に近づけました(図3)。低い周波数では、理論的予測と比較して、深いランダムな変動がリコールの低下につながる可能性があります。逆に、我々の解析では、低品質と定義される10.1%のゲノムのリコールが著しく不良であることが示されました。
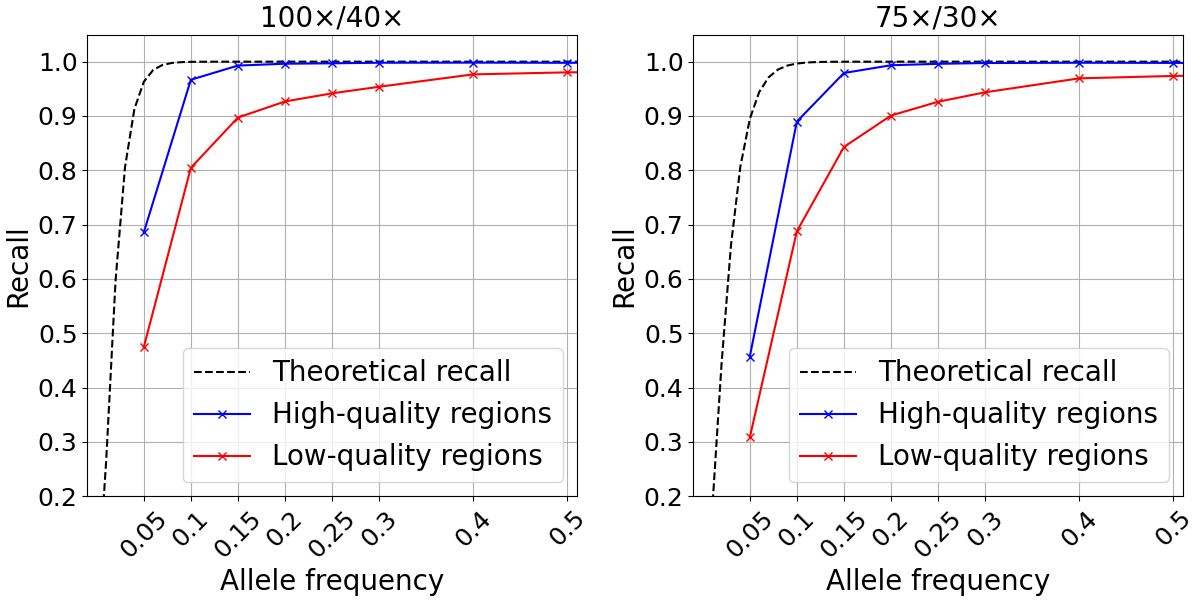
図3:高品質の領域(青色)と低い領域(赤色)の混合データセットにおける 体細胞SNPのリコール。混合シーケンス深度の組み合わせは、100x/40x(左)と75x/30x(右)です。
5. 正常/正常減算による偽陽性コールの 定量化
次に、体細胞サブトラクションで腫瘍と正常の両方と同じサンプルを使用して偽陽性率を定量化しました。腫瘍サンプルの真のバリアントは正常サンプルにも存在するため、結果として生じるコールは偽陽性です。正常/正常のサブトラクションでは、ゲノム全体の偽陽性SNPのレベルが低いことが示されました(100×/40×で274、75×/30×で196)(図4)。全体的な頻度は低いものの、偽陽性は系統的品質の低い領域に集まっています。具体的には、偽陽性SNVの86~89%が低品質領域で発見されました。つまり、すべての低品質領域を除外するシンプルなフィルターは、真の陽性を約10%削減するコストでほとんどの誤検出を除去することになります。
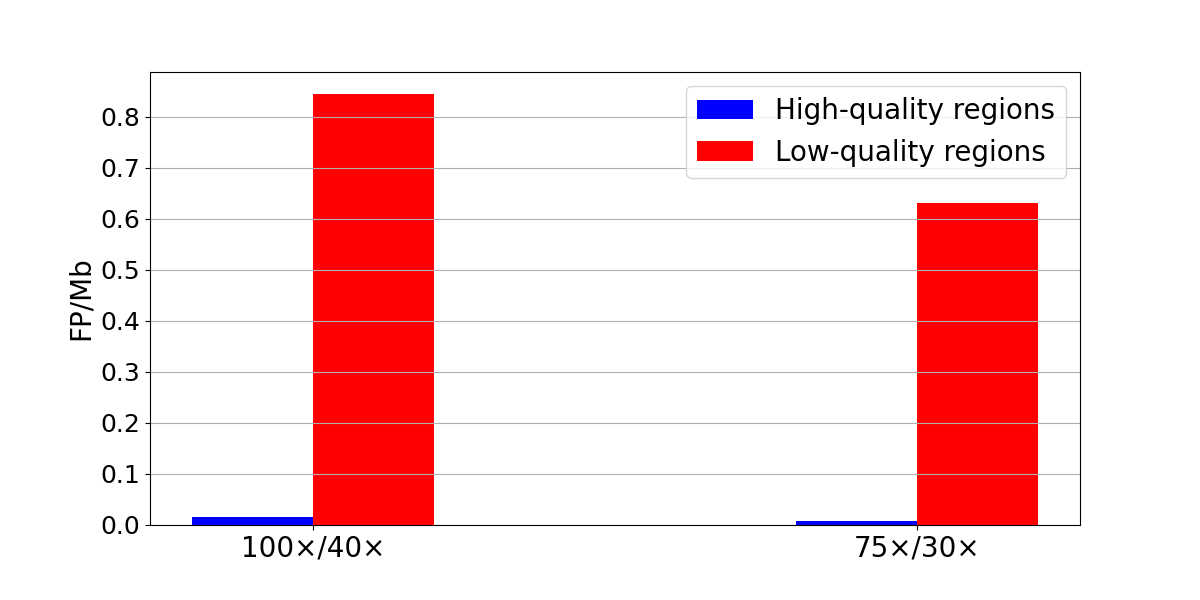
図4: 正常/正常サブトラクションでは、低品質領域で236の偽陽性SNP、100x/40x(左)で38の高品質領域で38の偽陽性SNP、低品質領域で176の偽陽性SNP、100x/40x(右)で高品質領域で20の偽陽性SNPが得られます。偽陽性の密度は、偽陽性SNPの数をMbの領域のサイズで割ったものとして計算されます。
ディスカッション
経験的に定義された系統的品質を用いることで、高いシーケンス性能を特徴とするゲノム領域の効果的な同定が可能になります。ここでは、これらの領域も、系統的品質が低い領域に対して高いリコールと低い偽陽性密度を特徴とすることを実証しました。
また、高い系統的品質の領域で達成されたリコール性能は、両方の試験深度の理論上の限界に近似していることも示しています。これは、ディープシーケンスが、低品質領域で低頻度バリアントを自信を持って呼び出すための唯一の戦略である可能性を示唆しています。逆に、ゲノム位置の89.9%を占める系統的品質の高い領域では、100×および75×シーケンスにより、15%を超えるアリル頻度と低い偽陽性率を持つバリアントの高いレベルのリコールが保証されます。
参考文献
- Bekritsky MA, Colombo C, Eberle MA. 高品質の単一ヌクレオチドバリアントコーリングによるゲノム領域の同定。イルミナのウェブサイト。 2022年2月16日にアクセス。
- Eberle MA, Fritzilas E, Krusche P, et al. 3世代の17員の家系のシーケンスから遺伝によって検証された540万のフェーズドヒトバリアントのリファレンスデータセット。 Genome Res. 2017;27(1):157-164. doi:10.1101/gr.210500.116
- Illumina. 腫瘍/正常研究における体細胞変異コーリングの評価。 2014年3月21日発行。2022年2月16日にアクセス。
- Illumina. DRAGEN体細胞。 2022年2月16日にアクセス。
- Real Time Genomics. RTGツール。 2022年2月16日にアクセス。