ヒトの体はおよそ250の細胞タイプで構成されており、それぞれが特定の遺伝子発現パターンによって定義されます。1RNAアトラスは、正常状態の細胞または組織のトランスクリプトームシグネチャーをカタログ化し、疾患の潜在的なバイオマーカーを定義することを目的としています。この目標に向けた研究の実績には、Human Cell Atlas、Genotype-Tissue Expression(GTEx)プロジェクト、Functional ANnoTation Of the Mammalian genome project(FANTOM5)、The Cancer Genome Atlas(TCGA)プログラムなどがあります。2-7しかし、既存のコンソーシアムデータは不完全です。今日までに、GTEx、FANTOM5、TCGAはポリアデニル化(polyA)メッセンジャーRNA(mRNA)または低分子RNAのいずれか、および特定の組織または疾患状態に焦点を当ててきました。
Human Cell Atlasに関連する研究では、シングルセルRNAシーケンス(scRNA-Seq)と空間的RNA-Seq手法を使用して、さまざまなヒト組織を構成する個々の細胞タイプをマッピングし、新しい希少な細胞タイプを発見しました。8-12 シングルセル遺伝子発現は、不均一なサンプルで貴重な細胞ごとの分離能を提供します。 しかし、各細胞のトランスクリプトームの限られた視野しかありません。13、14 典型的なscRNA-Seq実験では、 3,000~6,000の転写産物、 少なくとも1つまたは2つのタグで表され、 セルごとに検出されます。この少ない転写産物で細胞を表現型解析できますが、最も発現率の高いmRNAのみを明らかにすることができます。特定の細胞型におけるノンコーディングRNA(ncRNA)の種類と量に関する情報が欠落している。トランスクリプトームをより詳細に把握するには、より包括的なシーケンスが必要です。
トランスクリプトーム百科事典の重要性
イルミナのサイエンティストと共同研究者は、究極のRNAアトラスを組み立てるには、より包括的で包括的な手法が必要であると考えていました。深く徹底したトランスクリプトーム百科事典は、scRNA-Seqデータを解釈し、がんやその他の疾患の起源組織を同定する研究者にとって、基本的なリソースとなる可能性があります。
個々の細胞タイプのディープシーケンス
ゲント大学の共同研究者を通じて、当社の統合研究チームは、個々の細胞型の160の 均質なコレクションにアクセスできました。15 これらの精製された細胞集団は、シングルセルシーケンスに焦点を当てたバルクRNAシーケンス深度の利点を可能にしました。同じ細胞特異的RNAサンプルで複数のアッセイを実行する能力により、豊かなトランスクリプトームの複雑さが明らかになり、確率的遺伝子発現からのバックグラウンドノイズの解決が容易になりました。また、45 の異なる組織と93の 細胞株をシーケンスし、そのうち89は13の 異なる種類のがんに由来するがん細胞株でした。約300種の 異なる組織と細胞タイプの各サンプルを非常に深くシーケンスし、それらの細胞に発現するほぼすべての転写産物を示しました(図 1)。
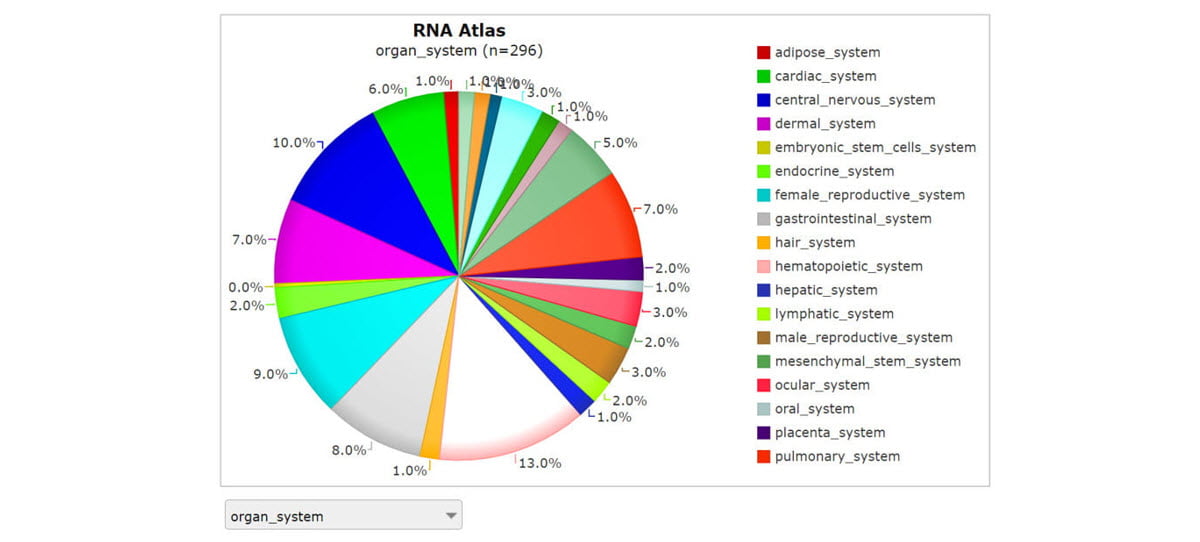
R2のスクリーンショット:ゲノミクス解析および可視化プラットフォーム。包括的なRNAアトラス研究では、3つの相補的なライブラリー調製アプローチを用いて、約300の異なる組織と細胞タイプに対して鎖特異的RNA-Seqを実施しました。そのアプローチは、total RNA-Seq、polyA RNA-Seq、およびsmall RNA-Seqです。
トランスクリプトーム解析に対する包括的なアプローチ
この研究では、3つの相補的なRNA-Seqライブラリー調製アプローチ(図 2)を利用して、完全なトランスクリプトームを調べました。
- リボソームRNA(rRNA)が枯渇したストランド型トータルRNA
- polyA転写産物の捕捉による鎖mRNA
- マイクロRNA(miRNA)やその他の小さなncRNAを調べるためのストランド型スモールRNA
追加のライブラリー調製法であるRNAエクソームエンリッチメント(図 2)では、バイオフルードなどの低インプットサンプルからのターゲットRNA-Seqが可能になりました。16
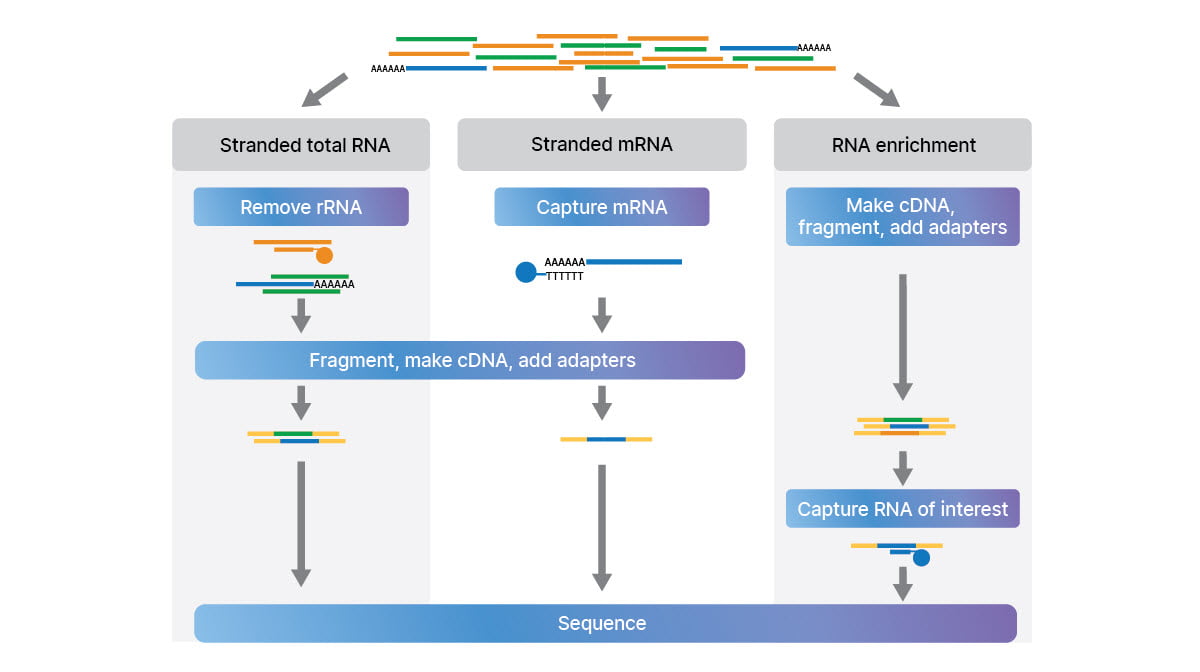
RNA-Seqライブラリー調製の相補的方法:rRNA枯渇を伴う鎖状全RNA、polyAキャプチャーを伴う鎖状mRNA、RNAエクソームエンリッチメント。低分子RNA法は示されていません。
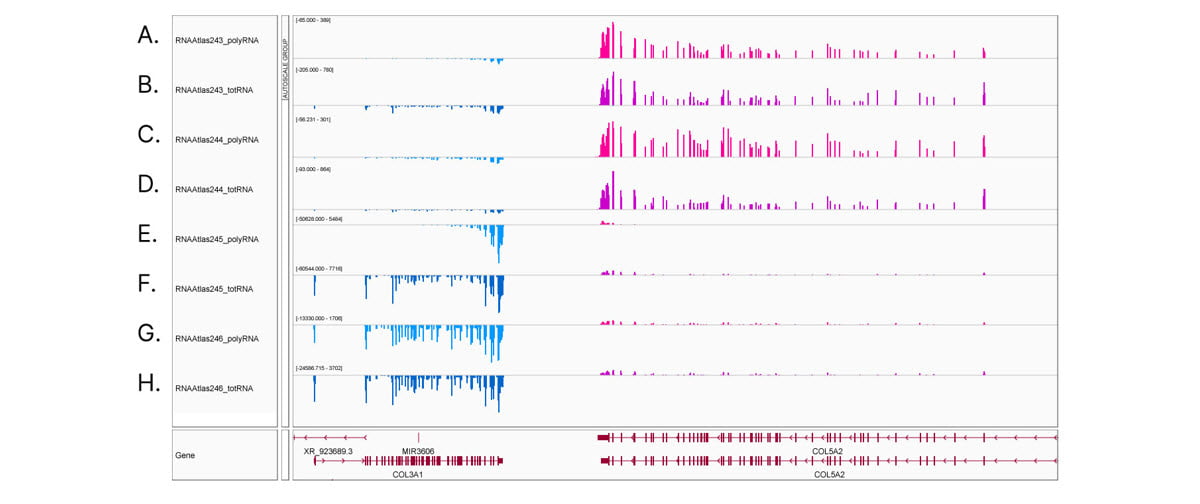
包括的で深いトランスクリプトーム解析は、鎖RNA-Seq法の重要性を示しています。眼細胞型におけるCOL3A1およびCOL5A2遺伝子発現のIntegrative Genomics Viewer(IGV)スクリーンショット。Watson鎖の転写産物は青色、Crick鎖の転写産物はピンク色です。(A,B)角膜上皮細胞、(C,D)ケラトサイト、(E,F)網膜色素上皮細胞、および(G,H)水晶体上皮細胞からの(A,C,E,G)鎖ポリA RNA-Seqおよび(B,D,F,H)鎖トータルRNA-Seq。各サンプルトラックの自動スケールで示されるリードカウント。角膜上皮細胞および角化細胞は、COL3A1の低発現とCOL5A2の高発現を示します。対照的に、網膜色素上皮細胞および水晶体上皮細胞は、COL3A1の高い発現とCOL5A2の低い発現を示します。網膜色素上皮細胞における(E)polyA RNA-Seqと(F)total RNA-Seqを比較すると、polyA RNA-Seqライブラリーに共通の3'バイアスが明らかになります。
新規転写産物の検索
RNAアトラストランスクリプトームは、ヒトRNAデータベースに対してアノテーションされ、既知と新規の両方の転写産物を同定しました。 比較データセットにはGENCODE、 RefSeq、 FANTOM5、 Comprehensive Human Expressed SequenceS(CHESS)、 MiTranscriptome、 BIGTトランスクリプトーム、 いくつかの小規模RNAおよびncRNAデータベースがあります。6、15、17-20 新規転写産物の独立した確認のため、 遺伝子発現のキャップ解析(CAGE)6とクロマチンプロファイルを介したプロモーターマッピングからの転写開始部位データを使用しました。21 このRNAアトラス研究で明らかになった新しい転写産物のほとんどはncRNAでした。 イントロンおよび遺伝子間miRNAおよび長鎖遺伝子間ncRNA(lincRNA)の予測値を含みます。15 Stranded RNA-Seqは、シングルエクソンlincRNAの有効性を確認する上で極めて重要でした。15
遺伝子制御における新しいパラダイムの解明
この包括的なRNAアトラスによって明らかになった1つの洞察は、ポリアデニル化に関する従来のドグマに課題を投げかけています。この研究では、lincRNAの75%以上が非ポリアデニル化されています(すなわち、ポリAが選択したRNA-Seqライブラリー調製よりも、全RNA-Seqに多く存在していました)。さらに、RNAアトラスの開発中、何千ものノンコーディング転写物が、組織特異的なポリアデニル化の差異パターンを示しました。15このポリアデニル化の差異の実際の意義は不明ですが、将来の研究では、遺伝子制御におけるその役割と疾患状態のバイオマーカーとしての使用の可能性を調べることができます。
ヒト生体液アトラス
容易にアクセス可能なバイオフルードを使用した早期発見と疾患の発生は、治療の選択肢と転帰に大きく影響します。ゲント大学の共同研究者とともに、唾液から汗、母乳まで、20の ヒトバイオフルードのRNAを調査しました。22興味深い発見の1つは、精液と涙液がRNAに豊富で、高品質のシーケンスライブラリーを生成したことでした。ヒトのバイオフルードは、循環RNA(circRNA)の豊富な供給源でもあり、22これらのRNAは、固有のバックスプライシングイベントによって形成され、例えばmiRNAまたは制御タンパク質に結合し、デコイ/スポンジとして機能することによって遺伝子発現の制御因子として機能することができます。その円形の性質により、ヌクレアーゼに対する耐性が高まり、生体液の安定性が向上し、潜在的なバイオマーカーとしての魅力が高まります。23,24
バイオフルードプロファイルは、それらを作り出す組織を主に反映しています。研究者は、包括的なRNAアトラスからの深い洞察を使用して、生体液からRNAを原産組織に戻すことができます。例えば、精液は前立腺細胞からのRNAが豊富であったため、血液よりも前立腺がんをスクリーニングするためのリキッドバイオプシーのより良いソースとなる可能性があります。涙液からのRNAは、目の健康に関する情報を提供する可能性があります。
結論
RNAライブラリー調製ソリューションのポートフォリオを活用して、包括的なヒトトランスクリプトームアトラスとヒトバイオフルードアトラスという2つの豊富なリソースを創出しました。15,22これらのアトラスは、大規模で慎重に解析されたデータセットを他の研究者の手に渡すことで、科学的発見を加速することができます。その後の研究では、これらのRNAアトラスデータセットをマイニングして、複数のRNAタイプの発現と制御に関するより深い洞察を得ることができます。
詳細はこちら
論文を読む:
- Lorenzi L, Chiu HS, Avila Cobos F, et al. The RNA Atlasは、ヒトノンコーディングRNAのカタログを拡張しています。 Nat Biotechnol . 2021;39(11):1453-1465. doi:10.1038/s41587-021-00936-1
- Hulstaert E, Morlion A, Avila Cobos F, et al. Charting Extracellular Transcriptomes in The Human Biofluid RNA Atlas. Cell Rep . 2020;33(13):108552. doi:10.1016/j.celrep.2020.108552
RNAアトラスデータを調べる:
R2上の専用アクセスポータル:ゲノミクス解析および可視化プラットフォーム
参考文献
- Hatano A, Chiba H, Moesa HA, et al. セルペディア:細胞研究と分化解析のためのヒト細胞情報のレポジトリ。 データベース(Oxford)。2011;2011:bar046。doi:10.1093/database/bar046
- Human Cell Atlas. humancellatlas.org/. 2022年2月15日にアクセス。
- Lindeboom RGH, Regev A, Teichmann SA. ヒト細胞アトラスに向けて:過去のメモを取る。 Trends Genet. 2021;37(7):625-630. doi:10.1016/j.tig.2021.03.007
- Rozenblatt-Rosen O, Shin JW, Rood JE, et al. 高品質のヒト細胞アトラスの構築。 Nat Biotechnol. 2021;39(2):149-153. doi:10.1038/s41587-020-00812-4
- Broad Institute of MIT and Harvard. Genotype-Tissue Expression (GTEx) project. gtexportal.org/home/. 2022年2月16日にアクセス。
- RIKEN. Functional annotation of the mammalian genome (FANTOM5) project. fantom.gsc.riken.jp/5/. 2022年2月16日にアクセス。
- National Cancer Institute. The Cancer Genome Atlas (TCGA) program. cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga. 2022年2月16日にアクセス。
- Wilbrey-Clark A, Roberts K, Teichmann SA. Cell Atlasのテクノロジーと組織構造への洞察。 Biochem J. 2020;477(8):1427-1442. doi:10.1042/BCJ20190341
- Deprez M, Zaragosi LE, Truchi M, et al. Human Healthy Airwaysのシングルセルアトラス。 Am J Respir Crit Care Med. 2020;202(12):1636-1645. doi:10.1164/rccm.201911-2199OC
- Luecken MD, Zaragosi LE, Madissoon E, et al. discovAIRプロジェクト:ヒト肺細胞アトラスへのロードマップ。 Eur Respir J. 2022;2102057. doi:10.1183/13993003.02057-2021
- Haniffa M, Taylor D, Linnarsson S, et al. ヒト発生細胞アトラスのロードマップ。 Nature. 2021;597(7875):196-205. doi:10.1038/s41586-021-03620-1
- Plasschaert LW, Žilionis R, Choo-Wing R, et al. 気道上皮の単細胞アトラスは、CFTRが豊富な肺のイオン細胞を明らかにします。 Nature. 2018;560(7718):377-381. doi:10.1038/s41586-018-0394-6
- Mereu E, Lafzi A, Moutinho C, et al. 細胞アトラスプロジェクトのためのシングルセルRNAシーケンスプロトコールのベンチマーキング。 Nat Biotechnol. 2020;38(6):747-755. doi:10.1038/s41587-020-0469-4
- Chen G, Ning B, Shi T. シングルセルRNA-Seqテクノロジーおよび関連するコンピューターデータ解析。 Front Genet. 2019;10:317. doi:10.3389/fgene.2019.00317
- Lorenzi L, Chiu HS, Avila Cobos F, et al. RNA Atlasは、ヒトノンコーディングRNAのカタログを拡張します。 Nat Biotechnol. 2021;39(11):1453-1465. doi:10.1038/s41587-021-00936-1
- Illumina. 循環転写産物の検出が改善されました。 2021年発行。2022年2月17日にアクセス。
- Frankish A, Diekhans M, Ferreira AM, et al. ヒトおよびマウスゲノムのGENCODEリファレンスアノテーション。 核酸 Res. 2019;47(D1):D766-D773. doi:10.1093/nar/gky955
- Frankish A, Diekhans M, Jungreis I, et al. GENCODE 2021。 核酸 Res. 2021;49(D1):D916-D923. doi:10.1093/nar/gkaa1087
- O'Leary NA, Wright MW, Brister JR, et al. NCBIのリファレンスシーケンス(RefSeq)データベース:現在のステータス、分類学的拡大、機能アノテーション。 核酸 Res. 2016;44(D1):D733-D745. doi:10.1093/nar/gkv1189
- Pertea M, Shumate A, Pertea G, et al. CHESS:数千もの大規模RNAシーケンス実験からキュレーションされた新しいヒト遺伝子カタログは、広範な転写ノイズを明らかにします。 Genome Biol. 2018;19(1):208. doi:10.1186/s13059-018-1590-2
- National Institutes of Health. NIH Roadmap Epigenomics Mapping Consortium. The Roadmap Epigenomics Project. roadmapepigenomics.org/. 2022年2月23日にアクセス。
- Hulstaert E, Morlion A, Avila Cobos F, et al. ヒト体液RNAアトラスにおける細胞外トランスクリプトームのチャート化 Cell Rep. 2020;33(13):108552. doi:10.1016/j.celrep.2020.108552
- Verduci L, Tarcitano E, Strano S, Yarden Y, Blandino G. CircRNA:ヒトの疾患における役割とバイオマーカーとしての使用の可能性。 Cell Death Dis. 2021;12(5):468. doi:10.1038/s41419-021-03743-3
- Li X, Yang L, Chen LL. 循環RNAの生物形成、機能、課題。 Mol Cell. 2018;71(3):428-442. doi:10.1016/j.molcel.2018.06.034