はじめに
何百万ものヒトゲノムとエクソームがシーケンスされていますが、疾患の原因となる変異を良性の遺伝子バリエーションと区別するのが困難なため、臨床適用は依然として限られています1,2。その適応度に有害な影響があるため、臨床的に意義のある遺伝的バリアントはヒト集団では極めてまれである傾向があります3。したがって、集団において高頻度に発生するバリアントを観察することは、良性であることを裏付ける強力なエビデンスであり2,4、消去法によって病原性変異を系統的に特定することができます。様々なヒト集団にわたる共通バリエーションを分析することは、良性変異を分類する効果的な戦略ですが5、現在のヒトにおける共通バリエーションの総量は限られています。リファレンスゲノムの7,000万を超える潜在的なミスセンスバリアントのうち、全体の集団対立遺伝子頻度が0.1%を超えるのは1,000分の1程度です5,6。
現代のヒト集団以外では、チンパンジーは次に近い現存種であり、99.4%のアミノ酸配列同一性を共有しています7。ヒトとチンパンジーにおけるタンパク質コード配列がほぼ同一であることは、チンパンジーのタンパク質コードバリアントに作用する自然淘汰が、ヒトの同一変異の適応度への影響をモデル化できる可能性も示唆しています。状態が同じ多型が2つの種の適応度に同様に影響を与える場合、チンパンジー集団における高い対立遺伝子頻度でのバリアントの存在は、ヒトにおいて良性の結果を示し、既知の良性バリアントのカタログを大幅に拡大するはずです。これにより、チンパンジーバリアントで検証する必要がある仮説が策定されます。
ヒト集団では、一般的な霊長類バリアントが良性である傾向があることを実証しました。トレーニングデータとして、ヒト以外の霊長類6種の集団シーケンスから何十万もの一般的なバリアントを使用して、病原性変異を高精度に予測するディープニューラルネットワークであるPrimateAIを開発しました。
他の霊長類によく見られるバリアントは、ヒトではおおむね良性
Exome Aggregation Consortium(ExAC)およびGenome Aggregation Database(gnomAD)で収集された123,136人のヒトで構成される集合エクソームデータが最近利用可能になったことで、対立遺伝子頻度スペクトル全体にわたるミスセンス変異および同義変異に対する自然選択の影響を測定することができます5。シングルトンバリアント(コホートで1回のみ観察)は、交絡因子で調整した後、de novo変異によって予測される2.2:1のミスセンス:同義比と近く一致しますが(図1a)8、対立遺伝子頻度が高いほど、自然選択による有害な変異の除去により、観察されたミスセンスバリアントの数が減少します。
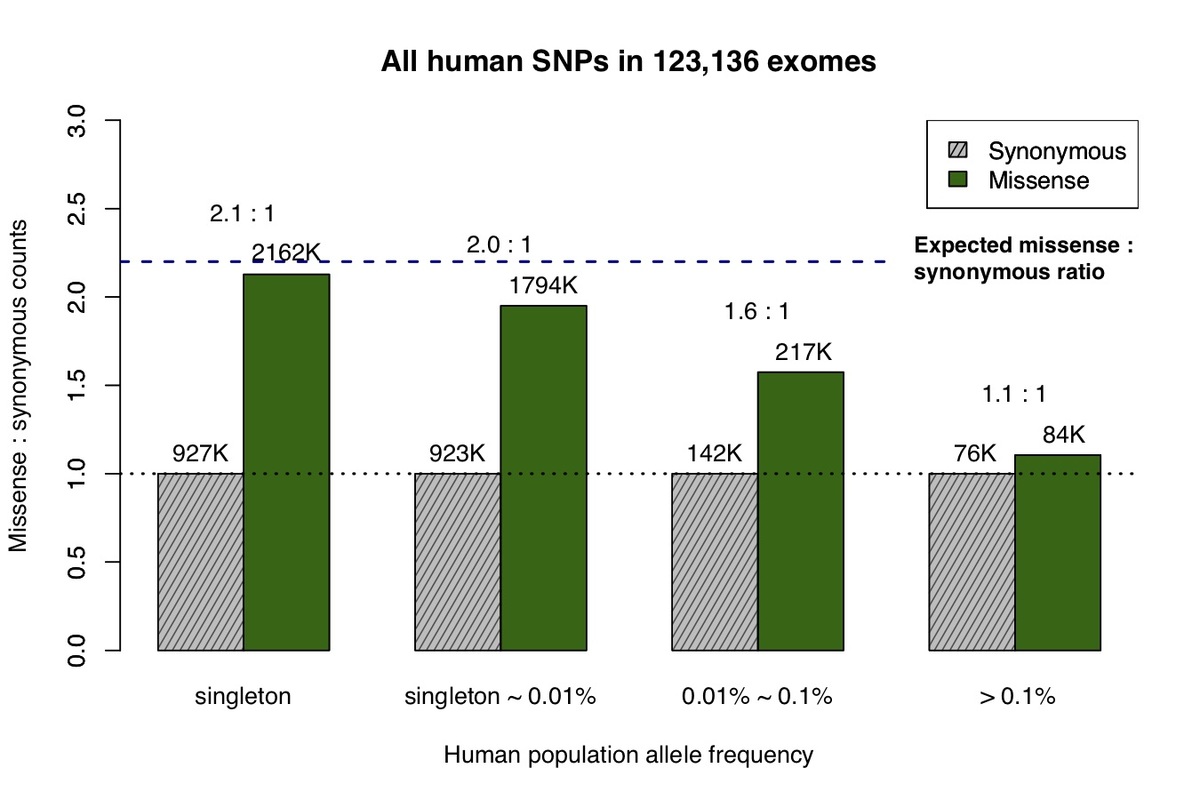
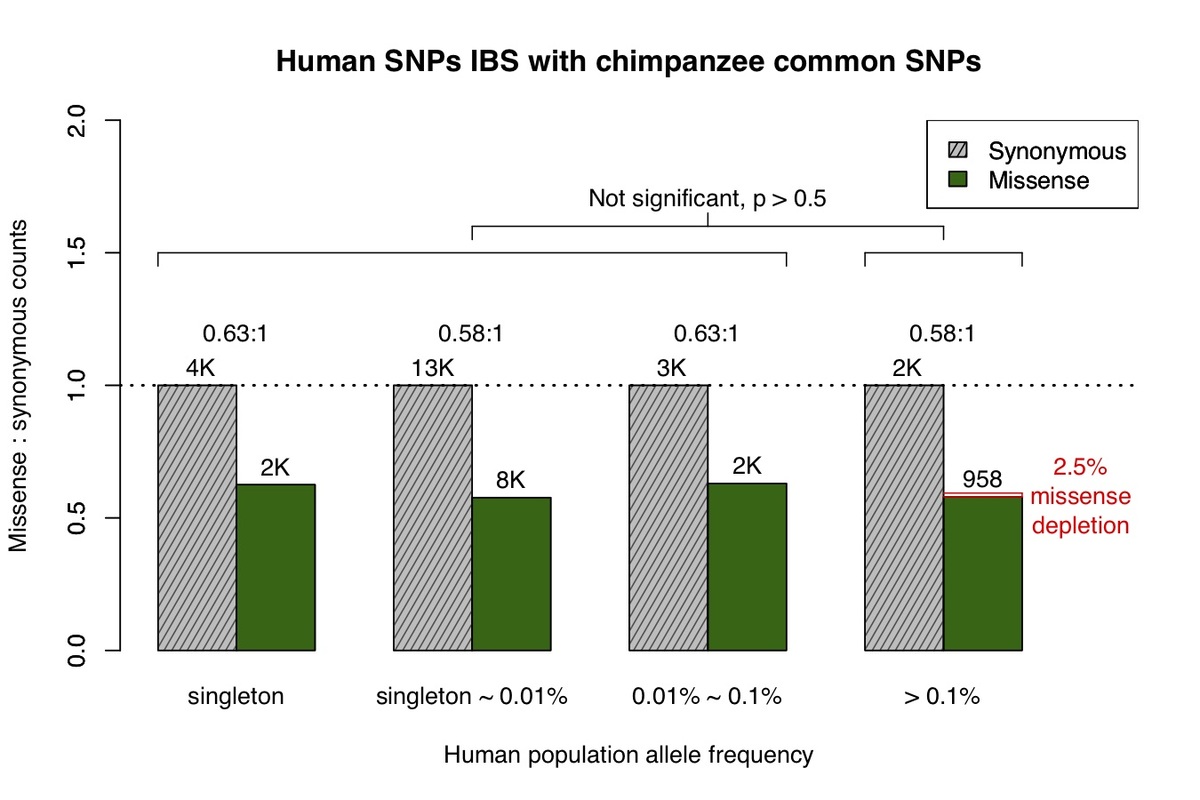
a、ExAC/gnomADデータベースで観察されたすべてのミスセンスおよび同義バリアントを、対立遺伝子頻度によって4つのカテゴリーに分けました。灰色の網掛けバーは各カテゴリーの同義バリアントの数を表し、濃い緑色のバーはミスセンスバリアントを表します。各バーの高さは、各対立遺伝子頻度カテゴリーの同義バリアント数に合わせて調整されます。b、チンパンジー共通バリアントと状態が同じ(IBS)ヒトミスセンスおよび同義バリアントの対立遺伝子頻度スペクトル。
霊長類バリアントは、great apeゲノムシーケンスプロジェクトおよびdbSNP9,10から取得されました。最初に、ヒトバリアントと状態が同じ一般的なチンパンジーバリアントを調べ(図1b)、ミスセンス:同義の比率がヒト対立遺伝子頻度スペクトル全体でほぼ一定であることを発見しました。これは、ヒト集団において一般的なチンパンジーバリアントに対するネガティブな選択がないことと一致しています。一般的なチンパンジーバリアントと状態が同じヒトバリアントで観察される低いミスセンス:同義の比率は、チンパンジーにおけるより大きな有効集団サイズと一致しており、これにより軽度に有害なバリエーションをより効率的にフィルタリングすることができます11,12。
次に、6つの非ヒト霊長類種のうち少なくとも1つでバリエーションが観察された、状態が同じヒトバリアントを特定しました。6種のそれぞれのバリエーションは、シーケンスされた個体数が限られていること、および各種で観察されたミスセンス:同義比が低いことに基づく一般的なバリアントを主に表しています。チンパンジーと同様に、ヒト以外の6種の霊長類のバリアントのミスセンス:同義比は、ヒトの対立遺伝子頻度スペクトル全体でほぼ同等であることがわかりました。ただし、一般的な対立遺伝子頻度でミスセンスバリエーションが軽度に除去されたこと(図2)は例外で、これは少数の希少バリアントが含まれているためと予想されます。
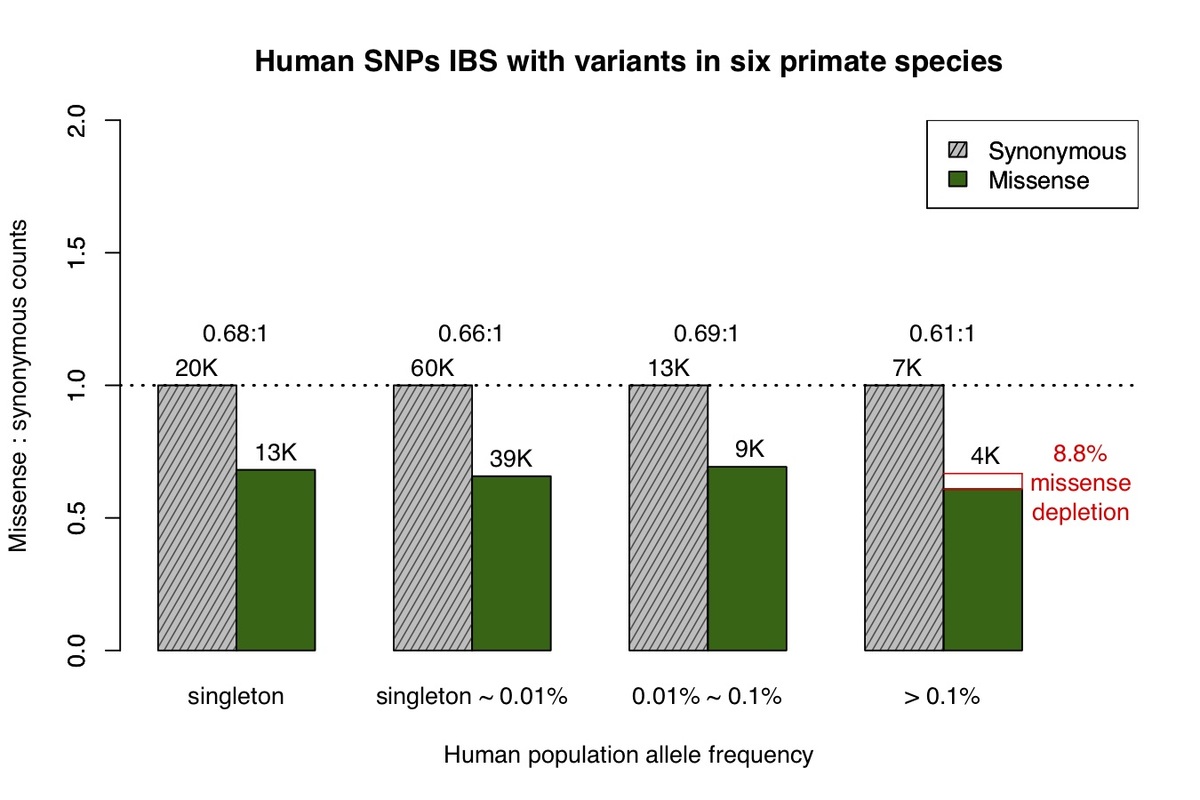
図2. 図1と同様に、ヒト以外の霊長類種の少なくとも1つで観察されるヒトミスセンスおよび同義バリアントの対立遺伝子スペクトル。
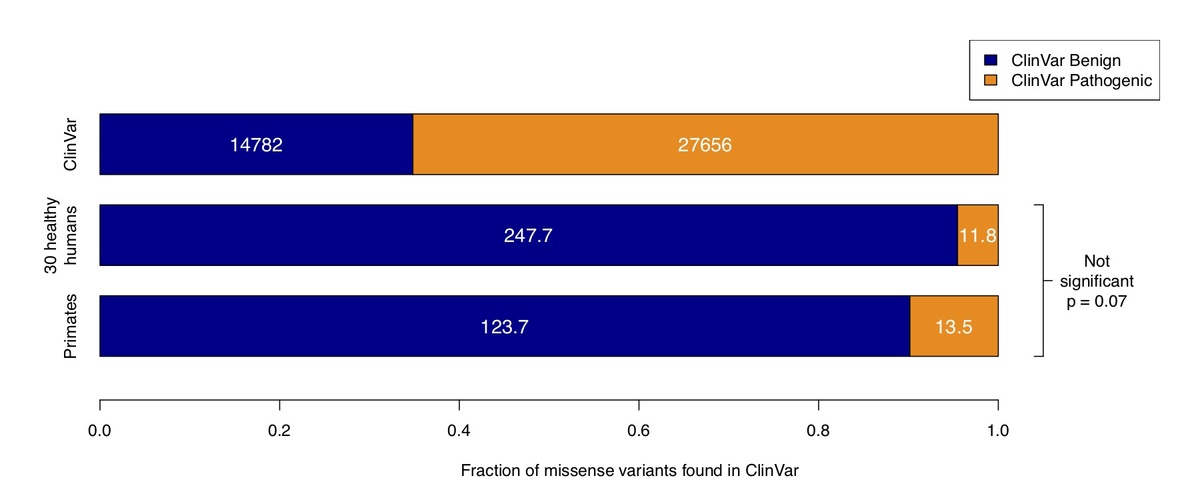
図3:ClinVarデータベース全体における良性および病原性のミスセンスバリアント数(上段)、ExAC/gnomAD対立遺伝子頻度からサンプリングしてシミュレーションしたヒト30例のコホートのClinVarバリアント(中段)、霊長類で観察されたバリアント(下段)の比較。矛盾する良性および病原性のアサーションと、重要性が不明な場合にのみアノテーションされたバリアントは除外されました。
バリアント病原性分類のためのディープラーニングネットワーク
臨床アプリケーションに対するバリアント分類の重要性は、この問題に対処するために教師あり機械学習を使用する多くの試みにインスピレーションを与えてきましたが、トレーニング用の良性および病原性バリアントが確実にラベル付けされた適切なサイズの真実のデータセットがないことが、これらの取り組みの妨げとなっています14-24。ヒト専門家がキュレーションしたバリアントの既存のデータベースがカバーしているのは、ゲノムのごく一部であり、ClinVarデータベースのバリアントの約50%はわずか200の遺伝子(ヒトタンパク質コード遺伝子の約1%)から来ています。さらに、系統的研究では、多くのヒト専門家によるアノテーションは裏付けとなるエビデンスが疑わしいことが示されており5,25、1人の患者にしか観察されないまれなバリアントを解釈することの難しさが強調されています。ヒトの解釈バイアスを減らすため、最近の分類器は、一般的なヒト多型またはヒトとチンパンジーの固定置換についてトレーニングを受けています26-29が、これらの分類器は、ヒトがキュレーションしたデータベースでトレーニングされた以前の分類器の予測スコアも入力として用いています。これらのさまざまな手法の性能の客観的ベンチマーキングは、独立したバイアスのない真実のデータセットがない場合には困難でした30。
6種のヒト以外の霊長類(チンパンジー、ボノボ、ゴリラ、オランウータン、アカゲザル、マーモセット)に由来するバリエーションは、一般的なヒトのバリエーションと重複しない30万を超える固有のミスセンス変異をもたらし、浄化選択のふるいを通った良性の結果をもつ一般的なバリアントを主に表しており、機械学習アプローチで利用可能なトレーニングデータセットを大幅に拡大します。平均すると、各霊長類種は5万件のバリアントに相当する量をもたらし、それはClinVarデータベース全体の現在の合計数よりも多くのバリアントです。さらに、このコンテンツには人間の解釈におけるバイアスが含まれていません。
一般的なヒトバリアントと霊長類のバリエーションからなるデータセットを用いて、新しい深層残差ネットワークであるPrimateAI(https://github.com/Illumina/PrimateAI)をトレーニングしました。これは、関心のあるバリアントに隣接するアミノ酸配列と、他の種におけるオルソログ配列のアライメントを入力として受け取ります(図4a)31。人間が設計した特徴を使用する既存の分類器とは異なり、当社のディープラーニングネットワークは一次配列から直接特徴を抽出する方法を学習します。タンパク質構造に関する情報を組み込むため、個別のネットワークをトレーニングしてシーケンスのみから二次構造と溶媒露出度を予測し32,33、フルモデルにサブネットワークとして含めました(図4b)。結晶化に成功したヒトタンパク質の数が少ないことを考えると、一次配列から構造を推測することは、タンパク質構造が不完全であることや機能的ドメインアノテーションによるバイアスを回避するという利点があります。タンパク質構造を含むネットワークの総深度は、約400,000のトレーニング可能なパラメーターで構成される36の畳み込み層でした。
良性ラベルのバリアントのみを使用して分類器をトレーニングするために、特定の変異が集団の一般的なバリアントとして観察される可能性が高いかどうかとして予測問題を構成しました。いくつかの要因が、高い対立遺伝子頻度でバリアントを観察する確度に影響を与えます。このうち、有害性にのみ関心があります。良性トレーニングセットの各バリアントをラベルなしのミスセンス変異と対応付け、交絡因子を制御し、良性バリアントと対応付けられた対照群とを区別するようにディープラーニングネットワークをトレーニングしました8。ラベル付けされていないバリアントの数がラベル付けされた良性トレーニングデータセットのサイズを大きく上回るため、良性トレーニングデータセットに対応付けられたラベル付けされていないバリアントの異なるセットをそれぞれ使用する8つのネットワークを並行してトレーニングし、コンセンサス予測を得ました。
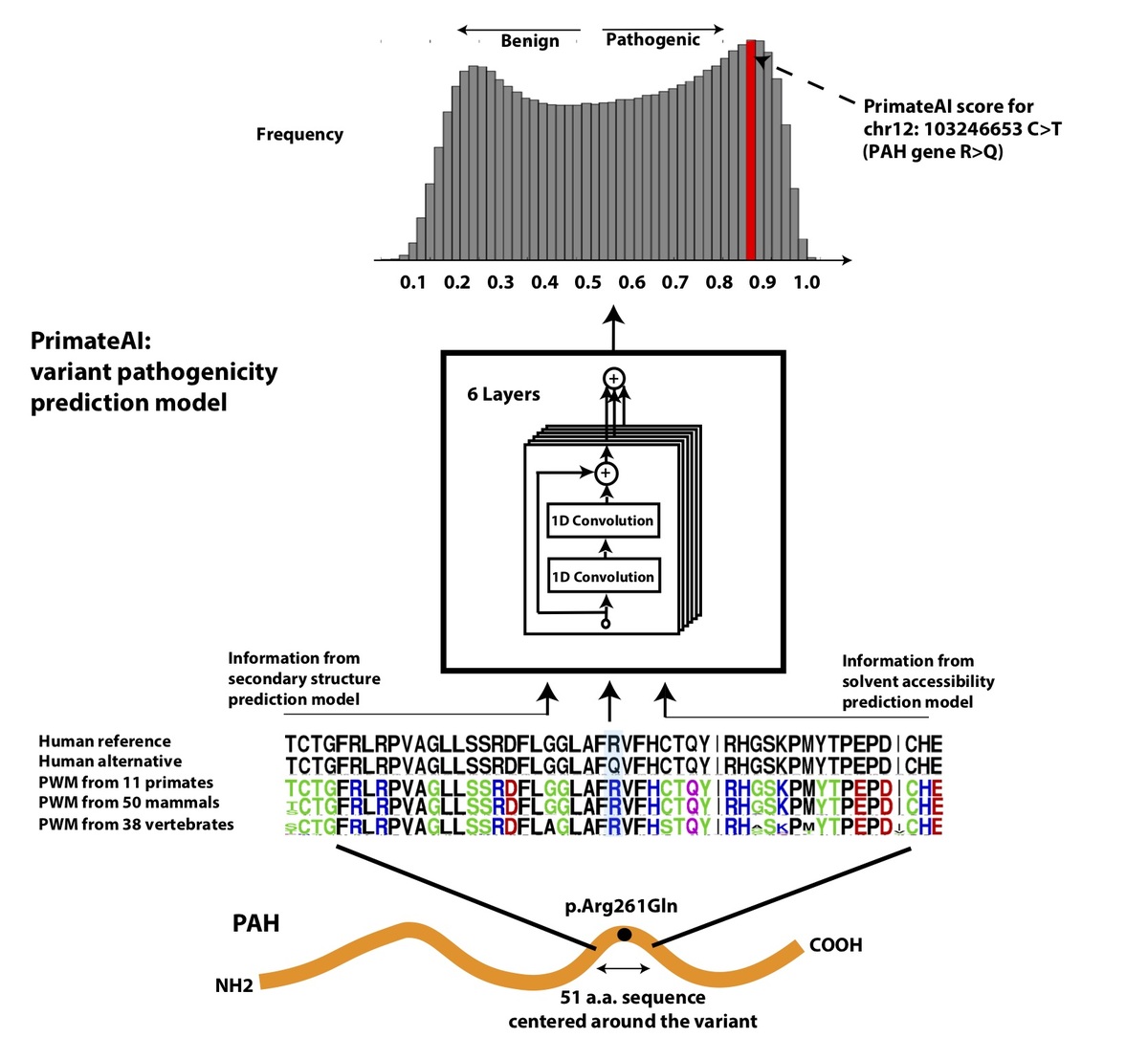
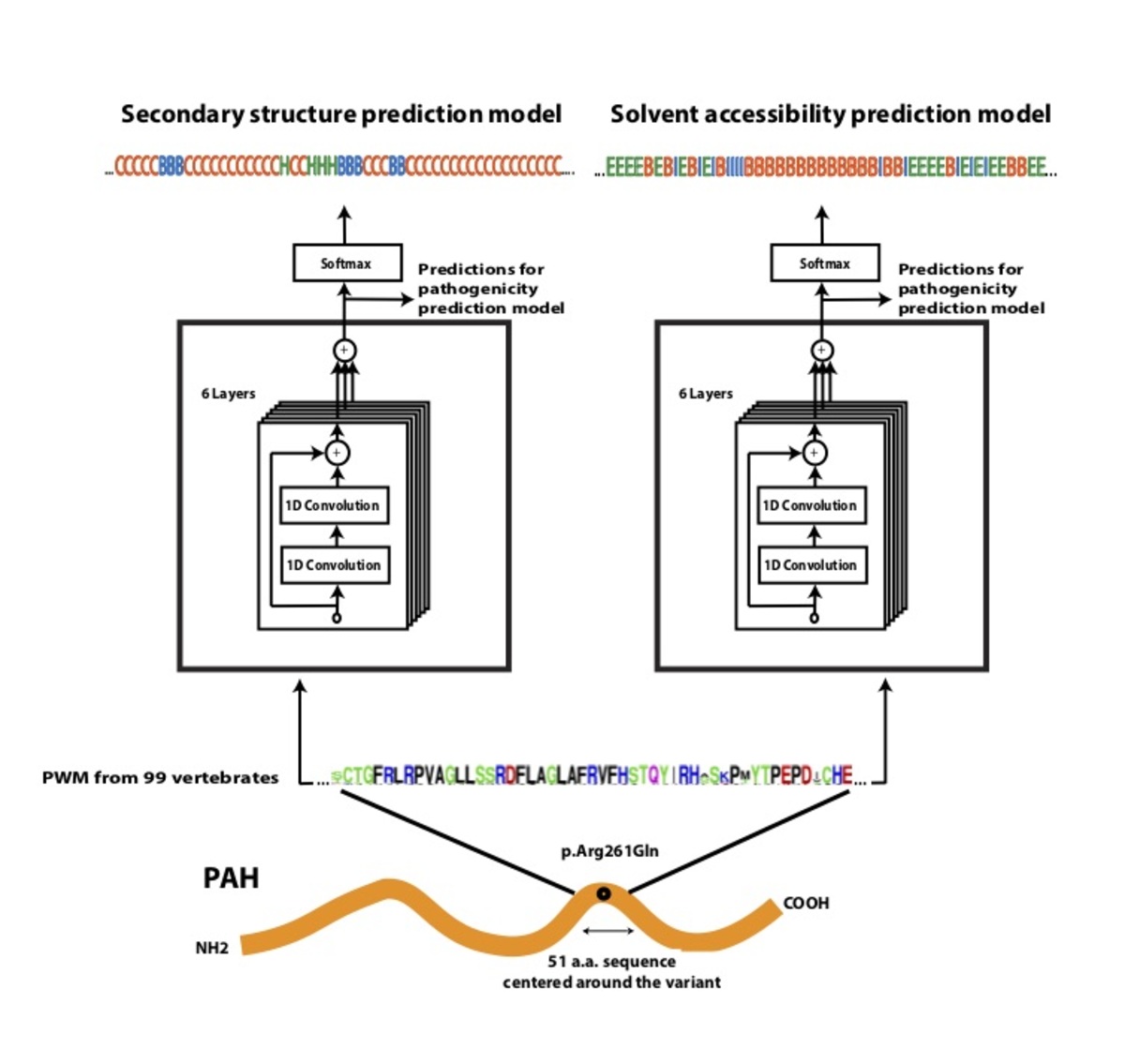
図4:病原性予測のためのディープラーニングネットワークであるPrimateAIのアーキテクチャ。 a、病原性予測のためのディープラーニングネットワークのアーキテクチャであるPrimateAI。PrimateAIスコアとして示される予測病原性は、0(良性)~1(病原性)の尺度で示されます。ネットワークは、バリアントを中心とするヒトアミノ酸(AA)リファレンスおよび代替シーケンス(51 AA)の入力として、99の脊椎動物種から計算された位置重みマトリックス(PWM)保存プロファイル、および b、二次構造および溶媒露出度予測ディープラーニングネットワークの出力を受け取ります。これらは、3状態タンパク質二次構造(ヘリックス—H、ベータシート—B、コイル—C)および3状態溶媒露出度(埋没—B、中間—I、露出—E)を予測します。
病原性予測の例
ディープラーニングネットワークは、一次アミノ酸配列のみをインプットとして使用し、てんかん、自閉症、知的障害の主要な疾患遺伝子である電位依存性ナトリウムチャネルSCN2A(図5)について示すように、重要なタンパク質機能ドメインの残基に高い病原性スコアを正確に割り当てます。SCN2Aの構造は4つの相同リピートで構成されており、それぞれに6つの膜貫通型ヘリックス(S1-S6)が含まれています34,35。膜が脱分極すると、正に荷電したS4膜貫通ヘリックスが膜の細胞外側に向かって移動し、S5/S6細孔形成ドメインがS4-S5リンカーを介して開くようになります。S4、S4-S5リンカー、S5ドメインの変異は、早期発症てんかん性脳症と臨床的に関連しており36、ネットワークにより、遺伝子の病原性スコアが最も高いと予測され、健康な集団ではバリアントが乏しいことが示されています。
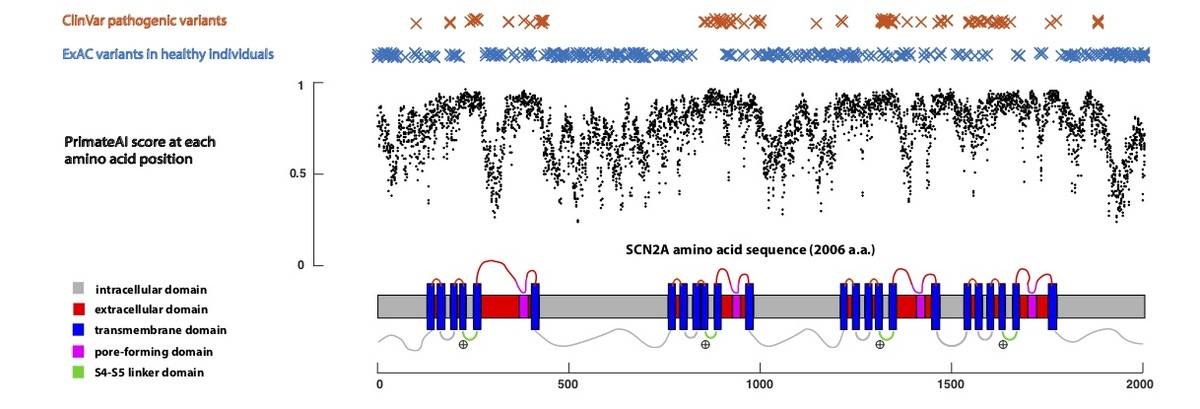
図5:SCN2A遺伝子の各アミノ酸位置における予測病原性スコア。主要な機能ドメインについてアノテーションされています。遺伝子に沿ってプロットされているのは、各アミノ酸位置におけるミスセンス置換の平均PrimateAIスコアです。
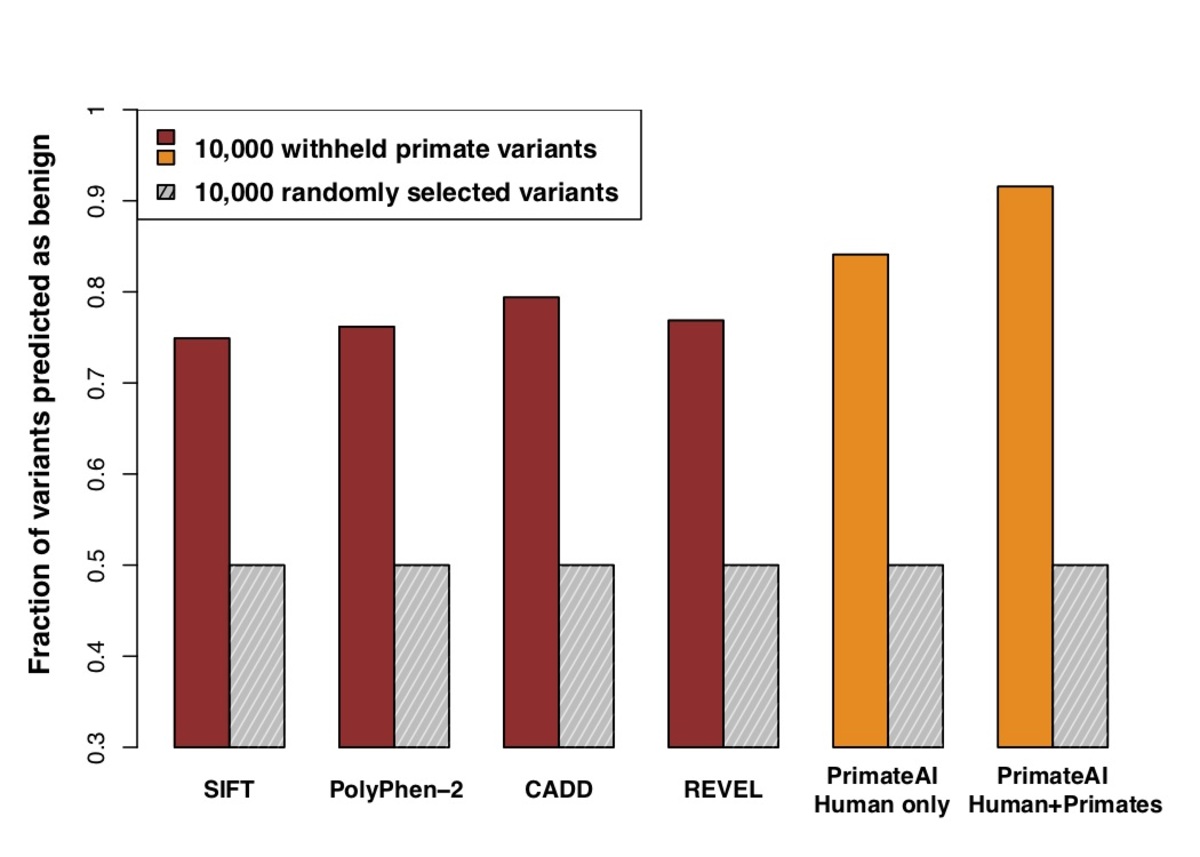
図6:トレーニングから除外した10,000の一般的な霊長類バリアントのテストセットにおける良性の影響予測についての分類器の比較。y軸は、変異率について対応付けた10,000のランダムバリアントのセットにおける各分類器の閾値を50パーセンタイルスコアに対して正規化した後、良性として正しく分類された霊長類バリアントの割合を示します。
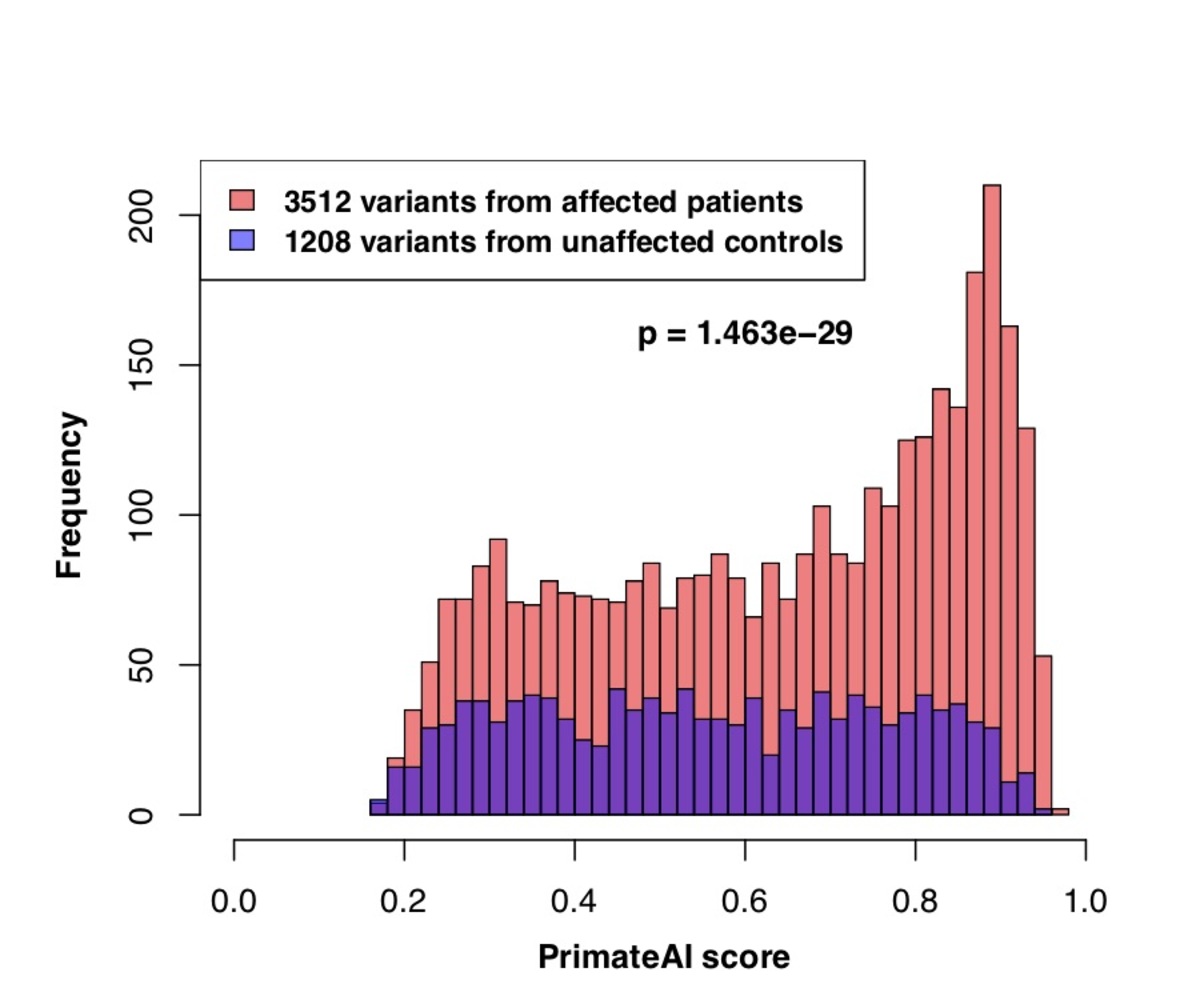
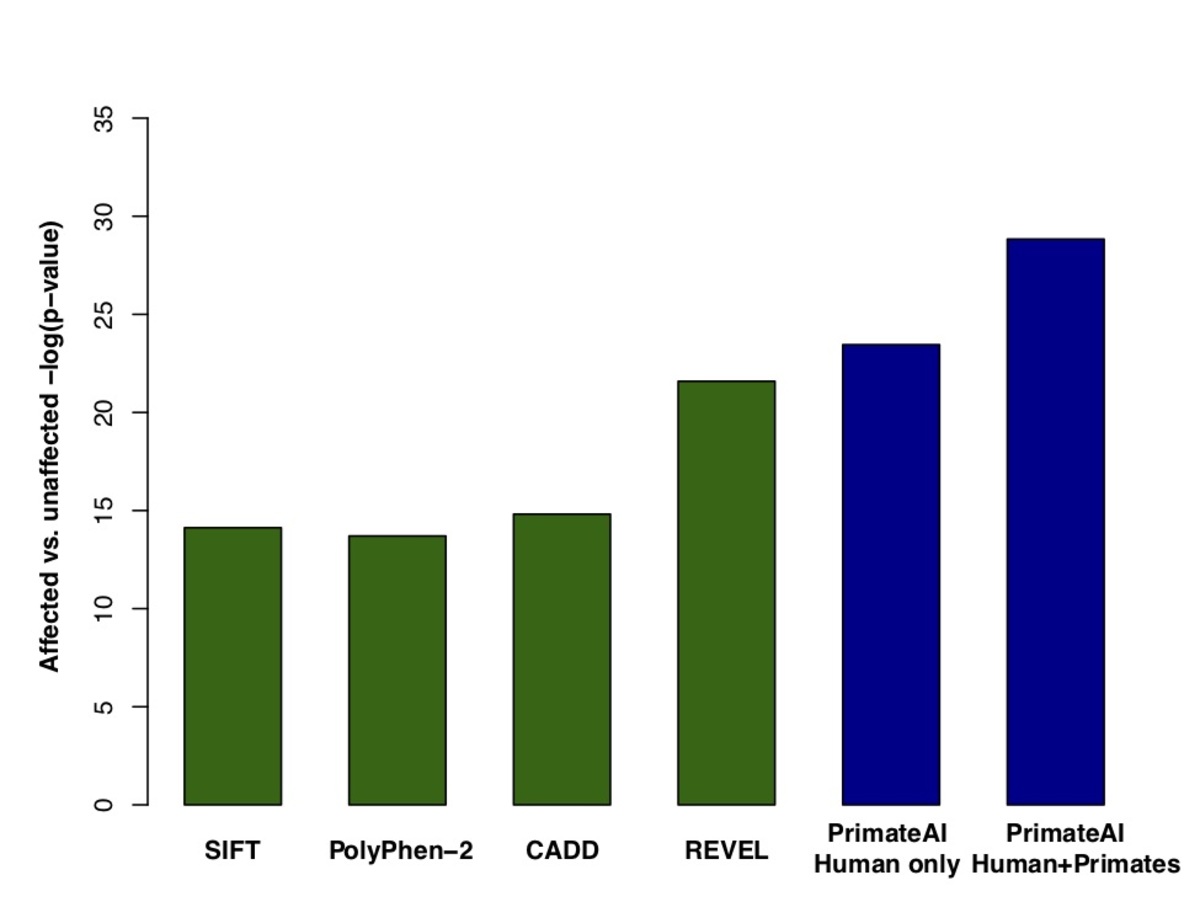
図7. a:影響を受けていない同胞と比較した、DDD患者で発生するde novoミスセンスバリアントのPrimateAI予測スコアの分布と、対応するWilcoxon順位和p値。 b:DDD症例と対照群におけるde novoミスセンスバリアントの分離に関する分類器の比較。Wilcoxon順位和検定のp値は、各分類器について示されています。
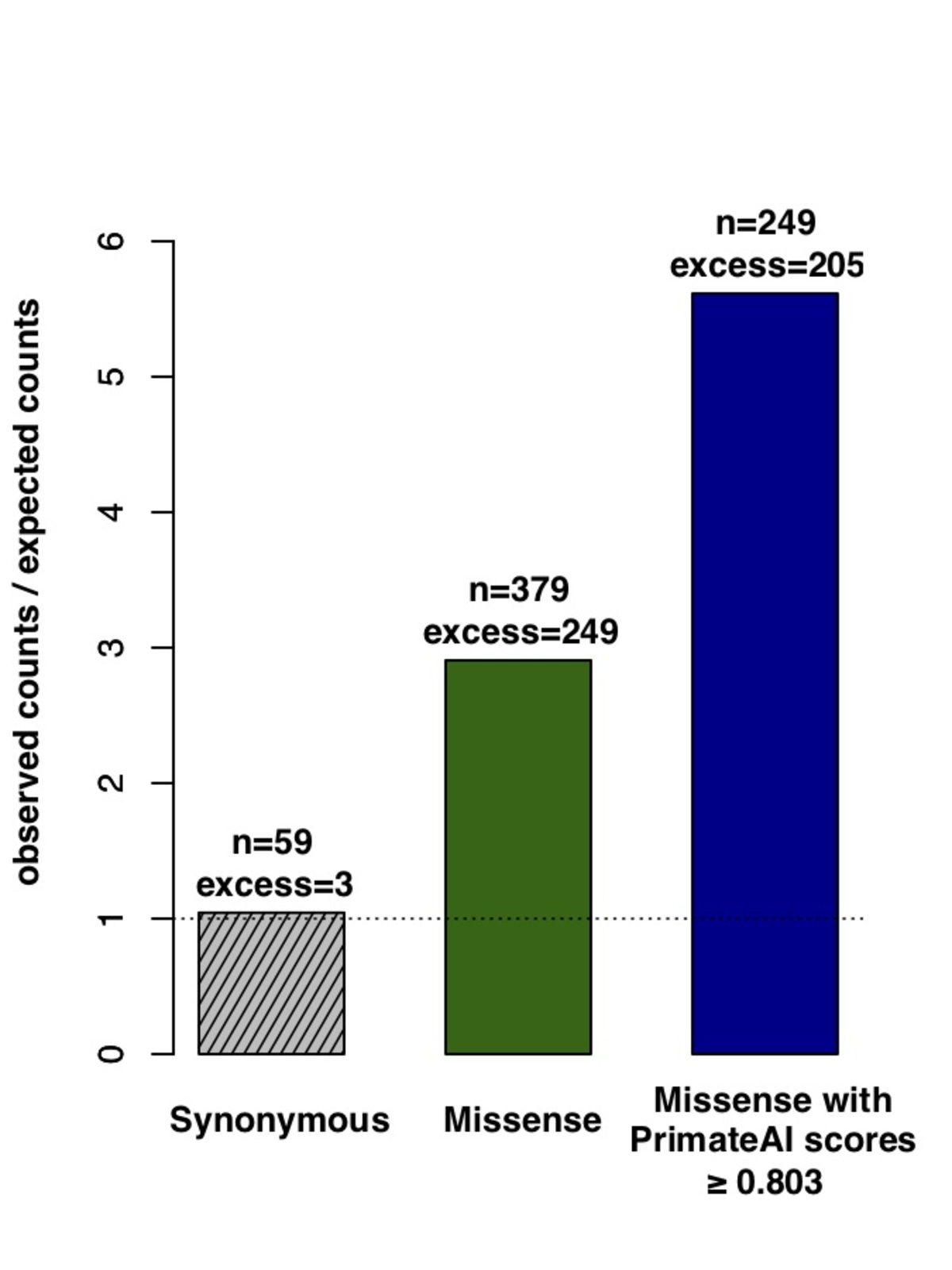
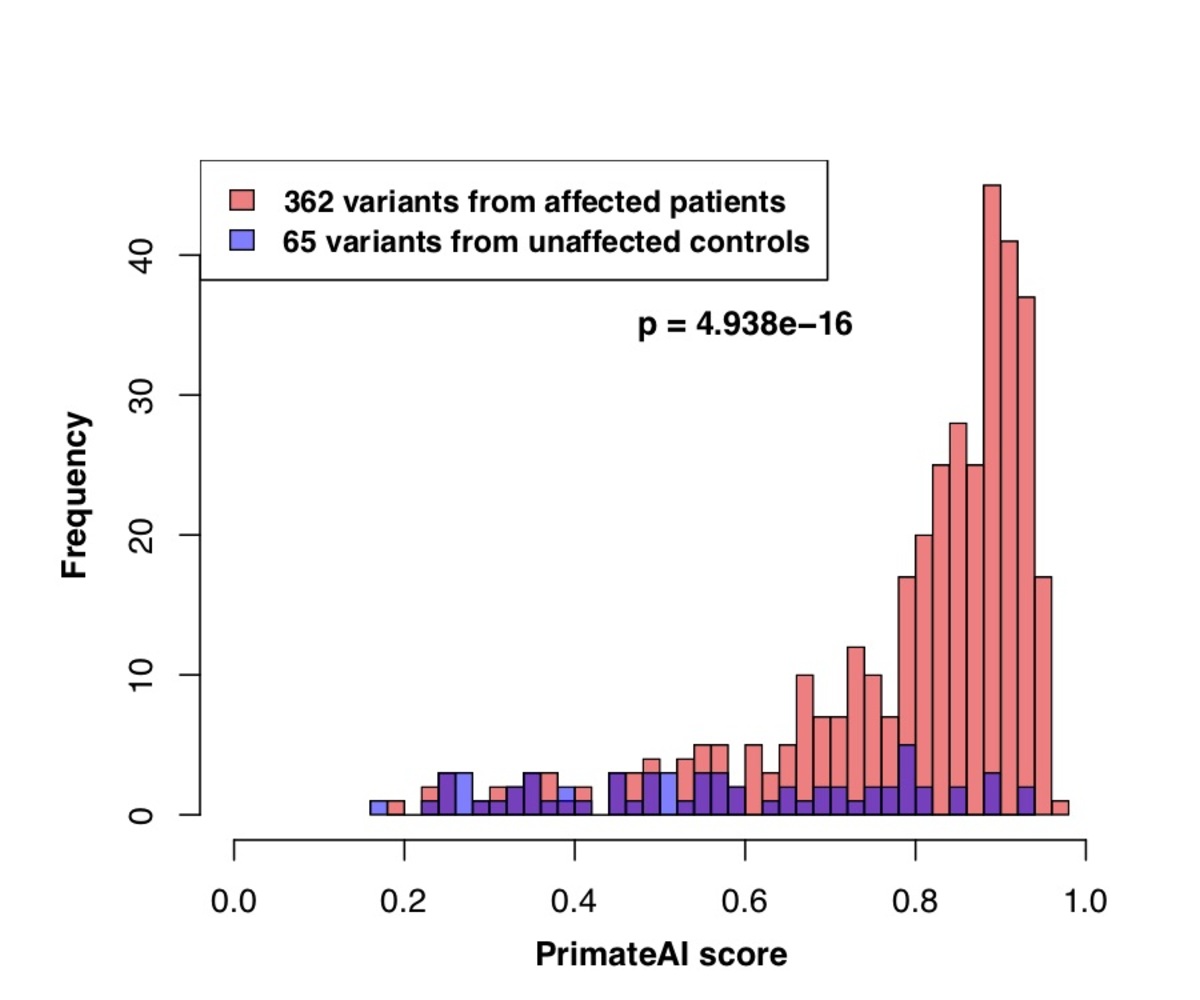
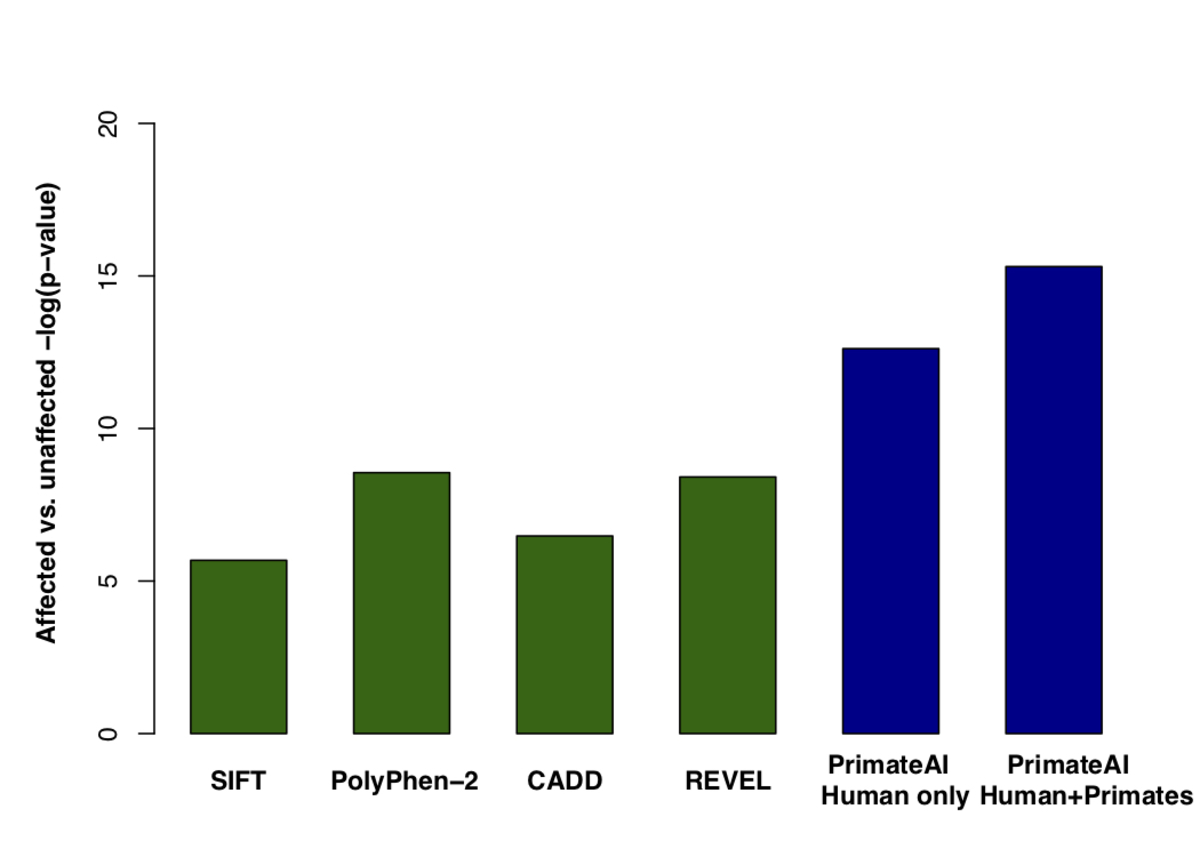
図8:P < 0.05の605 DDD遺伝子内の分類精度。a、de novoタンパク質切断バリエーションで有意(p<0.05)であった605の関連遺伝子内において、DDDコホートの罹患者で期待値を上回るde novoミスセンス変異のエンリッチメント。b、605の関連遺伝子内でDDD患者と非罹患の同胞に生じたde novoミスセンスバリアントのPrimateAI予測スコアの分布と、対応するWilcoxon順位和検定のp値。c、605遺伝子内で症例と対照群におけるde novoミスセンスバリアントを分離する各種分類器の比較。y軸は、各分類器のWilcoxon順位和検定のp値を示します。
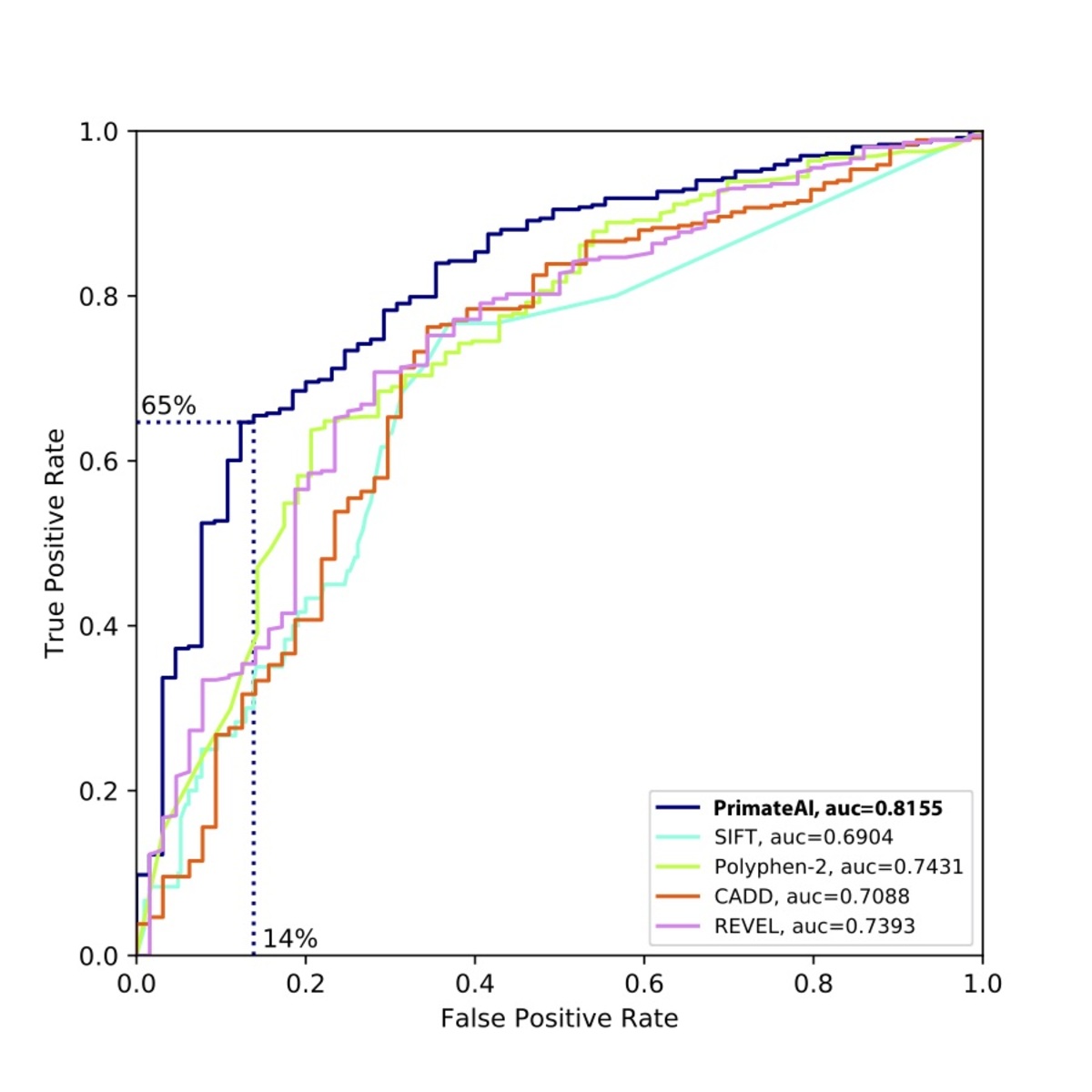
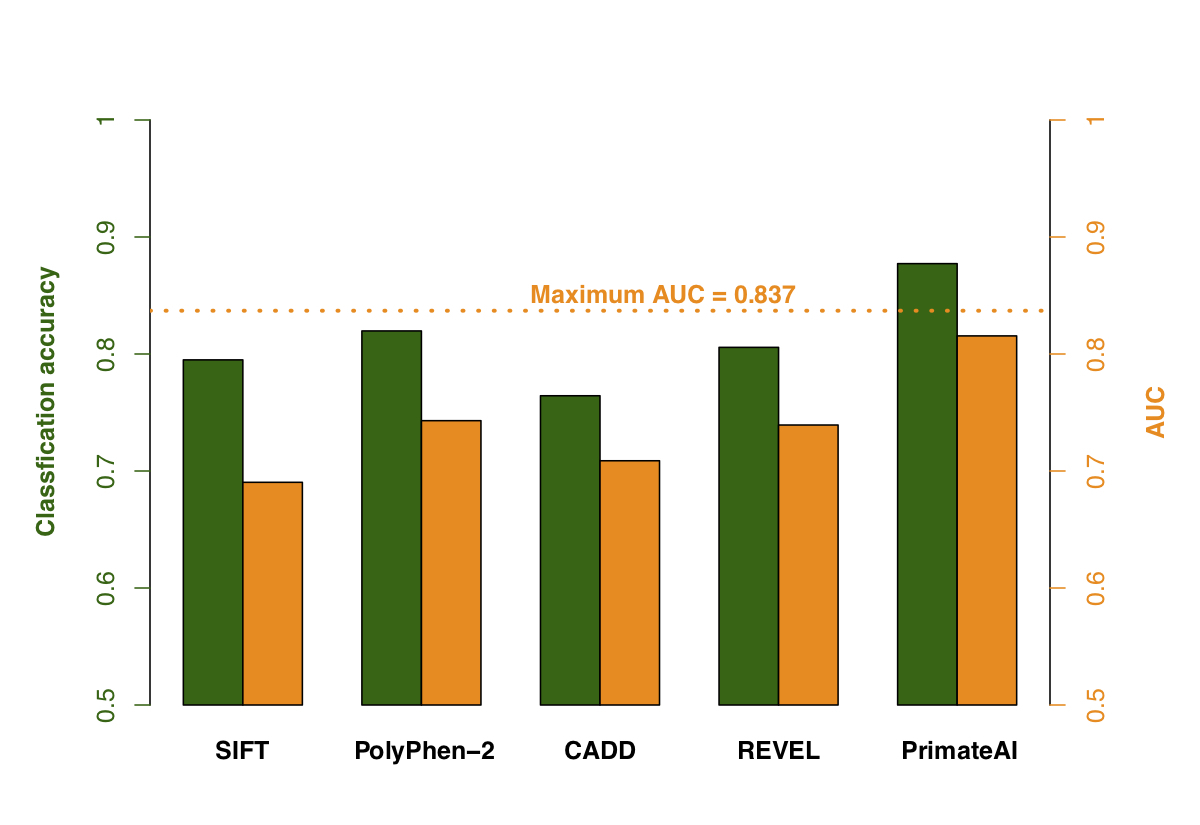
図9:さまざまな分類器の比較。a、受信者動作特性(ROC)曲線に示した性能で、各分類器の曲線下面積(AUC)を併記。b、各分類器の分類精度とAUC。示される分類精度は、真陽性と真陰性の誤り率の平均であり、図8aのエンリッチメントに基づく期待に従って、分類器が病原性と良性のバリアントを同数予測する閾値を用いています。DDD de novoミスセンスバリアントの33%がバックグラウンドを表すことを考慮し、完全な分類器で到達可能な最大AUCを点線で示しています。
当社の結果は、系統的な霊長類集団シーケンスが、現在臨床ゲノム解釈を制限している何百万もの意義不明のヒトバリアントを分類するための効果的な戦略であることを示唆しています。保留された一般的な霊長類バリアントと臨床バリアントの両方における当社のディープラーニングネットワークの精度は、ネットワークのトレーニングに使用される良性バリアントの数が増えるほど向上します。追加の霊長類種における一般的なバリエーションをカタログ化することで、意義不明の何百万ものバリアントの解釈が改善され、ヒトゲノムシーケンスの臨床的有用性がさらに向上します。
謝辞
J. K. Pritchard、M. E. Hurles、J. W. Belmont、およびR. E. Greenの洞察に富んだ議論に感謝いたします。Genome Aggregation Database(gnomAD)と、このリソースにエクソームおよびゲノムのバリアントデータを提供したグループに感謝いたします。Yanjun LiとXiaolin Liは、米国国立一般医学研究所および米国国立科学財団(助成金CNS-1747783、CNS-1624782、およびOAC-1229576)のR01GM110240によって部分的にサポートされました。Laksshman Sundaram、Samskruthi Reddy Padigepati、Jeremy F. McRae、Yanjun Li、Jack A. Kosmicki、Nondas Fritzilas、Jorg Hakenberg、Anindita Dutta、John Shon、Jinbo Xu、Serafim Batzloglou、およびXiaolin Liを含む原著論文の著者に謝意を表します。
外部リンク
Publication: https://pubmed.ncbi.nlm.nih.gov/30038395/
Software: https://github.com/Illumina/PrimateAI
Primate polymorphisms from the great ape genome project:
https://eichlerlab.gs.washington.edu/greatape/data.html
And from dbSNP database: https://www.ncbi.nlm.nih.gov/snp/
PrimateAI scores of 70 million variants: https://basespace.illumina.com/s/cPgCSmecvhb4
参考文献
- MacArthur, D. G. et al. Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469-476, doi:10.1038/nature13127 (2014).
- Rehm, H. L., J. S. Berg, L. D. Brooks, C. D. Bustamante, J. P. Evans, M. J. Landrum, D. H. Ledbetter, D. R. Maglott, C. L. Martin, R. L. Nussbaum, S. E. Plon, E. M. Ramos, S. T. Sherry, M. S. Watson. ClinGen--the Clinical Genome Resource. N. Engl. J. Med. 372, 2235-2242 (2015).
- Bamshad, M. J., S. B. Ng, A. W. Bigham, H. K. Tabor, M. J. Emond, D. A. Nickerson, J. Shendure. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 12, 745–755 (2011).
- Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405-424, doi:10.1038/gim.2015.30 (2015).
- Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285-291, doi:10.1038/nature19057 (2016).
- Liu, X., X. Jian, E. Boerwinkle. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. . Human Mutation 32, 894–899 (2011).
- Chimpanzee Sequencing Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69-87, doi:10.1038/nature04072 (2005).
- Samocha, K. E. et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet 46, 944-950, doi:10.1038/ng.3050 (2014).
- Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29, 308-311, doi:10.1093/nar/29.1.308 (2001).
- Prado-Martinez, J. et al. Great ape genome diversity and population history. Nature 499, 471-475 (2013).
- Kimura, M. The neutral theory of molecular evolution. Cambridge University Press, 1983
- de Manuel, M. et al. Chimpanzee genomic diversity reveals ancient admixture with bonobos. Science 354, 477-481, doi:10.1126/science.aag2602 (2016).
- Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44, D862-868, doi:10.1093/nar/gkv1222 (2016).
- Ng, P. C. & Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res 11, 863-874, doi:10.1101/gr.176601 (2001).
- Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248-249, doi:10.1038/nmeth0410-248 (2010).
- Chun, S., J. C. Fay. Identification of deleterious mutations within three human genomes. Genome Research 19, 1553-1561 (2009).
- Schwarz, J. M., C. Rödelsperger, M. Schuelke, D. Seelow. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576 (2010).
- Reva, B., Antipin, Y. & Sander, C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 39, e118, doi:10.1093/nar/gkr407 (2011).
- Dong, C. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 24, 2125-2137, doi:10.1093/hmg/ddu733 (2015).
- Carter, H., Douville, C., Stenson, P. D., Cooper, D. N. & Karchin, R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics 14 Suppl 3, S3, doi:10.1186/1471-2164-14-S3-S3 (2013).
- Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. PLoS One 7, e46688, doi:10.1371/journal.pone.0046688 (2012).
- Gulko, B., Hubisz, M. J., Gronau, I. & Siepel, A. A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat Genet 47, 276-283, doi:10.1038/ng.3196 (2015).
- Shihab, H. A. et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31, 1536-1543, doi:10.1093/bioinformatics/btv009 (2015).
- Quang, D., Chen, Y. & Xie, X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31, 761-763, doi:10.1093/bioinformatics/btu703 (2015).
- Bell, C. J., D. L. Dinwiddie, N. A. Miller, S. L. Hateley, E. E. Ganusova, J. Midge, R. J. Langley, L. Zhang, C. L. Lee, R. D. Schilkey, J. E. Woodward, H. E. Peckham, G. P. Schroth, R. W. Kim, S. F. Kingsmore. Comprehensive carrier testing for severe childhood recessive diseases by next generation sequencing. Sci. Transl. Med. 3, 65ra64(2011)。
- Kircher, M., D. M. Witten, P. Jain, B. J. O’Roak, G. M. Cooper, J. Shendure. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310-315 (2014).
- Smedley, D. et al. A Whole-Genome Analysis Framework for Effective Identification of Pathogenic Regulatory Variants in Mendelian Disease. Am J Hum Genet 99, 595-606, doi:10.1016/j.ajhg.2016.07.005 (2016).
- Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 99, 877-885, doi:10.1016/j.ajhg.2016.08.016 (2016).
- Jagadeesh, K. A., A. M. Wenger, M. J. Berger, H. Guturu, P. D. Stenson, D. N. Cooper, J. A. Bernstein, G. Bejerano. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nature Genetics 48, 1581-1586 (2016).
- Grimm, D. G. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Human Mutation 36, 513-523 (2015).
- He, K., X. Zhang, S. Ren, J. Sun. Proceedings of the IEEE conference on computer vision and pattern recognition. IEEE 770-778.
- Heffernan, R. et al. Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning. Sci Rep 5, 11476, doi:10.1038/srep11476 (2015).
- Wang, S., J. Peng, J. Ma, J. Xu. Protein secondary structure prediction using deep convolutional neural fields. Scientific Reports 6, 18962-18962 (2016).
- Payandeh, J., Scheuer, T., Zheng, N. & Catterall, W. A. The crystal structure of a voltage-gated sodium channel. https://www.nature.com/articles/nature10238
- Shen, H. et al. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. https://science.sciencemag.org/content/355/6328/eaal4326
- Nakamura, K. et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology 81, 992-998, doi:10.1212/WNL.0b013e3182a43e57 (2013).
- Vissers, L. E., Gilissen, C. & Veltman, J. A. Genetic studies in intellectual disability and related disorders. Nat Rev Genet 17, 9-18, doi:10.1038/nrg3999 (2016).
- Neale, B. M. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242-245, doi:10.1038/nature11011 (2012).
- Sanders, S. J. et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237-241, doi:10.1038/nature10945 (2012).
- De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209-215, doi:10.1038/nature13772 (2014).
- Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223-228, doi:10.1038/nature14135 (2015).
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433-438, doi:10.1038/nature21062 (2017).
- Iossifov, I. et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216-221, doi:10.1038/nature13908 (2014).
- Zhu, X., Need, A. C., Petrovski, S. & Goldstein, D. B. One gene, many neuropsychiatric disorders: lessons from Mendelian diseases. Nat Neurosci 17, 773-781, doi:10.1038/nn.3713 (2014).