はじめに
アルファサラセミアと脊髄性筋萎縮症(SMA)は、世界で最も蔓延している保因者の疾患です。それぞれの原因遺伝子であるHBA1/2およびSMN1における有害なコピー数バリアント(CNV)は、これらのキャリア状態の約95%を占めています。次世代シーケンサー(NGS)パネルデータは良好な結果を示しており、CNV検出に広く採用されていますが、HBA遺伝子とSMN遺伝子の両方は、擬似遺伝子やパラログとの相同性が高いため、一般的に標準シーケンスやバイオインフォマティクスワークフローではプロファイリングが難しいゲノムの非特異な領域で発生します。このような課題があるため、HBAおよびSMN遺伝子座のプロファイリングに特化したターゲットPCRベースの検査(MLPAやqPCRなど)は、キャリアスクリーニング研究で一般的に使用されるNGSパネルベースの検査を補完するためによく使用され、大幅な追加費用とワークフローの複雑さが増しています。
イルミナ全ゲノムシーケンス(WGS)データからHBA およびSMNを正確にジェノタイピングするターゲットバリアントコーラーが、それぞれDRAGEN v4.2およびv3.9でリリースされました。しかし、エクソームからのSMNとHBAの解析は、標準的なエクソームパネルがそのような発呼者で利用されるゲノム領域をカバーしなかったため、また、発呼者で当初利用された深度ノーマライゼーションアプローチでは、エンリッチメントベースのアッセイに固有のキャプチャー効率バイアスに対処するには不十分であったため、これまで不可能でした。
ここでは、DRAGEN v4.4でリリースされたSMNおよびHBAターゲットコーラーの更新版と、Illumina DNA Prep with Exome 2.5 Enrichmentパネルを補完するために使用できるスパイクインパネル(Illumina CS/PGx Custom Enrichment Research Panel)を提示し、イルミナエクソームの正確なHBAおよびSMNジェノタイピングを可能にします。
イルミナCS/PGxカスタムエンリッチメント研究パネル
多くのDRAGENターゲット発信者は、非エキソニック領域のカバレッジに依存しています。特に、SMN1/2コピー数コーリングに利用される一部の分化サイトと、HBAコピー数ジェノタイピングで使用される非相同領域は非エキソニックです。そのため、イルミナCS/PGxカスタムエンリッチメント研究パネルは、医学的に重要なさまざまな遺伝子のこのような領域を確実にカバーするために開発されました。スパイクインパネルでカバレッジが拡張された遺伝子を表1に示します。追加コンテンツの利用に必要なバイオインフォマティクスの改善は現在、HBAとSMNのみにリリースされていますが、イルミナCS/PGx Custom Enrichment Research Panelをエクソームパネルに追加することで、ユーザーはパネル内の他の遺伝子の今後のDRAGENリリースで計画されているバイオインフォマティクスの改善を活用できるようになります。
スパイクインパネルは合計359 kbのゲノムをカバーし、Illumina DNA Prep with Exome 2.5 Enrichmentパネルでカバーされるゲノムコンテンツの1%未満に相当します。したがって、エクソームパネルに追加した場合、スパイクインパネルは、固定レベルのサンプルマルチプレックスでエクソームターゲットのカバレッジの全体的なレベルに最小限の影響しか与えません。
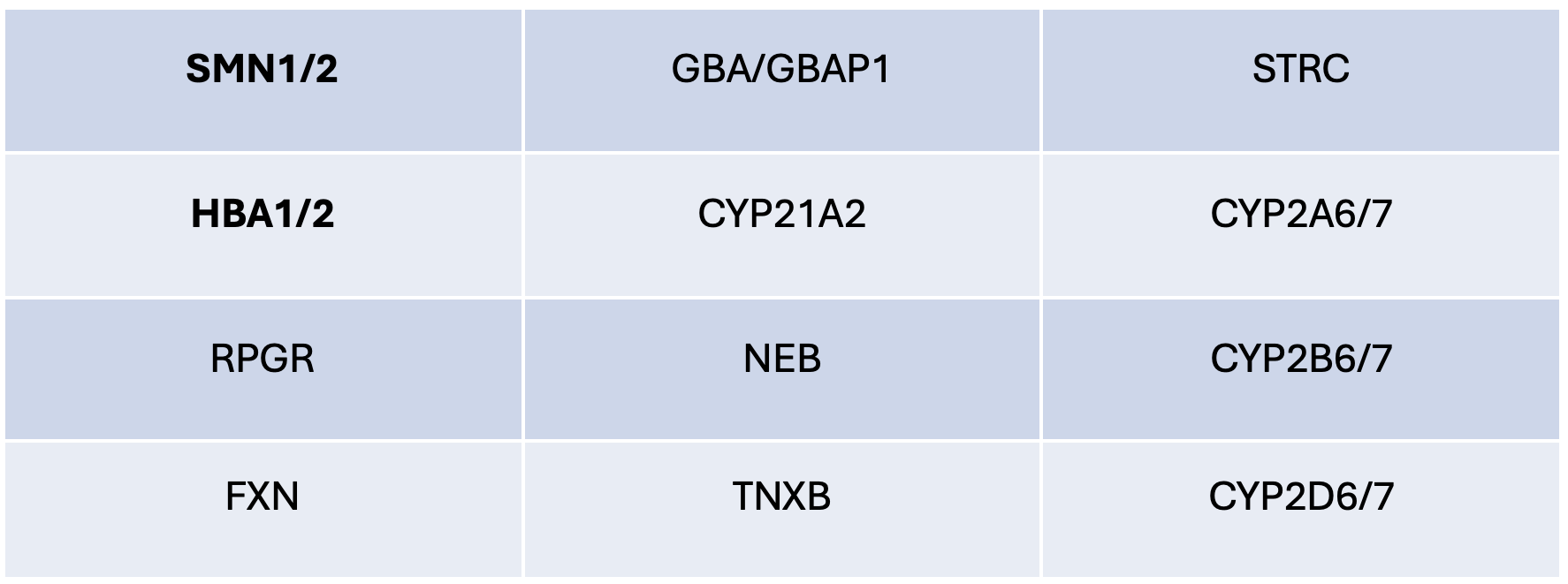
表1:スパイクインパネルのカバレッジが拡大した遺伝子のリスト
ターゲット発信者メソッドの概要
DRAGEN HBAコーラーは、HBA遺伝子座内およびHBA遺伝子座付近のいくつかの固有のサブ領域にわたってシーケンス深度シグナルを使用して、その遺伝子座に共通のコピー数遺伝子型の組み合わせを推測し、相同性媒介性の曖昧なリードアライメントの問題を克服します(図1A)。DRAGEN HBAコーラーは、αサラセミアの幅広い分子サブタイプをカバーするコピー数遺伝子型を検出できます。
DRAGEN SMNコーラー(図1B)は、いずれかの領域にアライメントされたリード数をカウントしてノーマライズすることでSMN1とSMN2の合計コピー数を特定します。エクソン1~6とエクソン7~8にまたがる領域については、共通のエクソン7~8の欠失を適切に処理するために、個別のコピー数推定が実施されます。次に、SMN1固有のコピー数を決定するために、異なるサイト間でスキャンが実行されます。SMN2コピー数は、合計コピー数からSMN1コピー数を差し引くことによって検出されます。
新しいエクソームベースのコーラーは、スパイクインプローブによって提供される拡張カバレッジと、ノーマルベースの深度ノーマライゼーション(バッチ内)のパネル、およびオリジナルのHBAとSMNコーラーの特殊なCNV検出技術を活用して、エクソームデータに対する正確なHBAとSMNジェノタイピングを可能にします(図1C)。
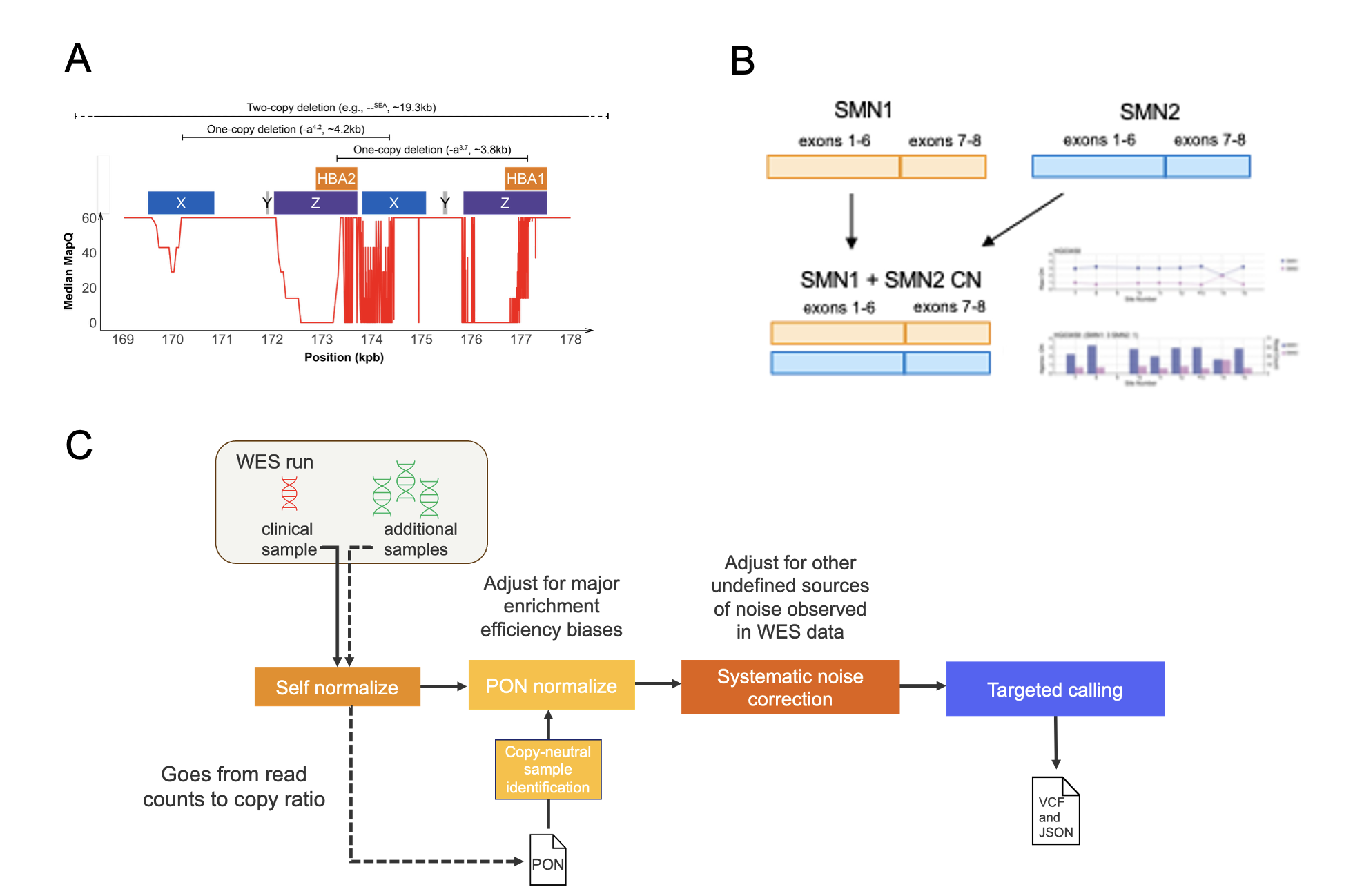
図1A:HBA1/2遺伝子座の特徴、その遺伝子座におけるリードのマッピング品質の中央値、およびその遺伝子座に頻繁に影響を与える病原性コピー数バリアントの表現。HBAコーラーは、固有のサブ領域を利用して、コピー数遺伝子型を推測します。
Figure1B:DRAGEN SMN1/2コピー数推定ワークフローの概略図
Figure1C:WESデータセットのHBAおよびSMNコーラーワークフローの概略図
内部細胞株コホートの結果
アッセイ開発中の精度を評価するために、エクソームパネルと添加スパイクインパネルを用いてイルミナでプロファイリングされた細胞株を使用しました。エクソームおよびスパイクインデータから行われたSMNおよびHBAコピー数ジェノタイプコールを、HBA(203のユニークサンプルから726のデータセット)およびSMN(216のユニークサンプルから739のデータセット)の直交的に確認されたコピー数ジェノタイプと比較して評価しました。本研究で使用した直交結果は、一致したWGSデータ上のDRAGENターゲットコーラー結果、または可能であればCoriellのqPCR/MLPAから導き出されました。3人の異なるオペレーターが手動ですべてのライブラリーを調製し、96のサンプル がS4フローセルにロードされたNovaSeq 6000システムでシーケンスしました。
更新されたターゲットコーラーを使用した全エクソームシーケンス(WES)から派生したHBAジェノタイプコールは、細胞株コホートにおける直交メソッドコールと100%(710/710)一致し、ノーコール率は2.2%でした。SMN1コピー数コールは、直交法と98.3%、SMAステータス分類と100%一致しました(影響ステータスはSMN1のコピーが0、保因ステータスはSMN1のコピーが1つ、影響ステータスはSMN1のコピーが2つ以上と定義)。WESからのSMN2コピー数コールは、直交法と99.3%一致していました。このコホートにおけるSMN1/2コピー数のノーコール率は1.1%でした。細胞株のアーチファクトにより、同一バッチ内の他のサンプルと大きく異なる全体的な深度プロファイルを持つサンプルによって、細胞株サンプルコホート全体で5.9%の全体的なQC失敗率が観察されました。
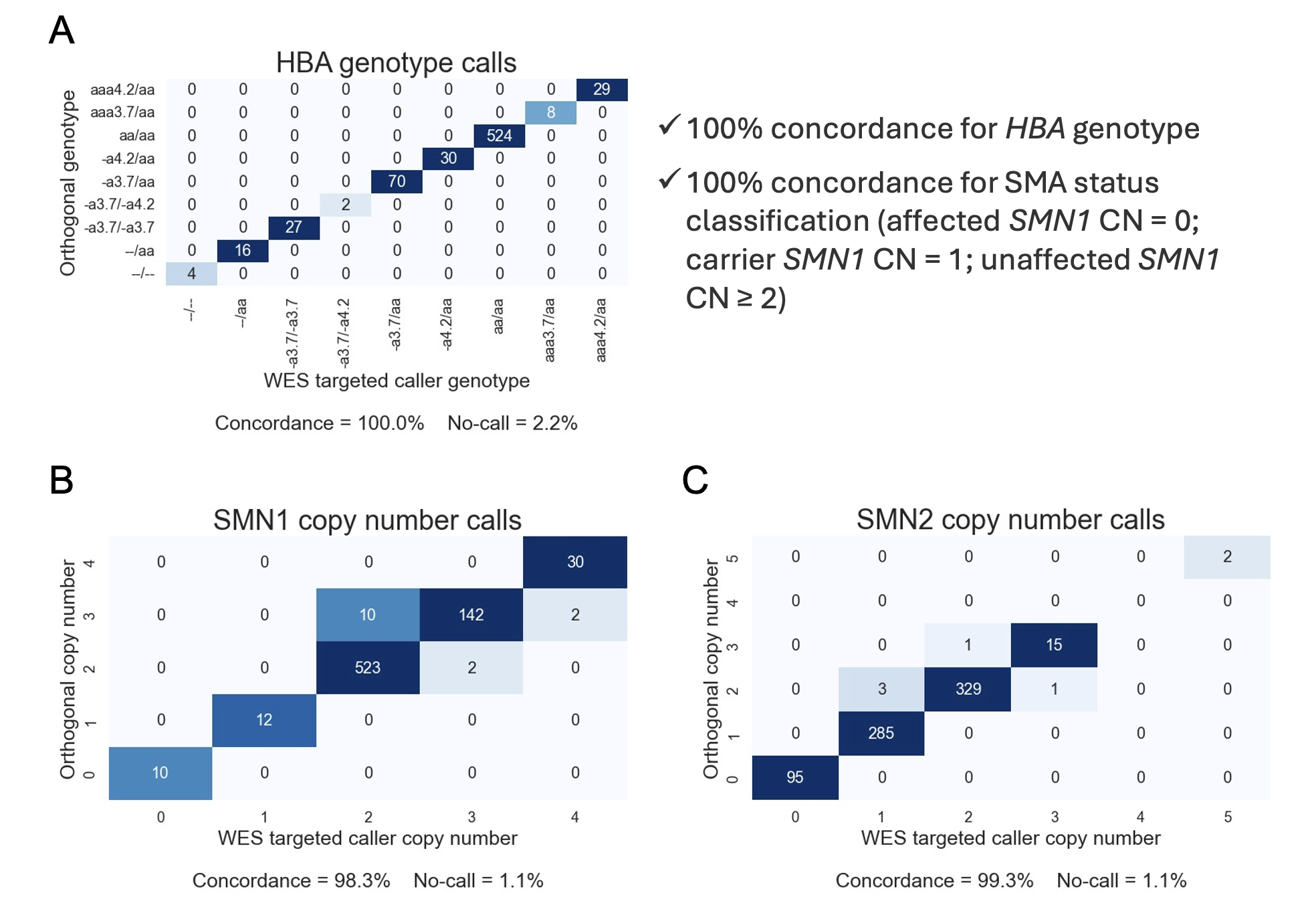
図1A:WESベースのHBAコーラーと、一致データセット上のWGSベースのHBAコーラーの結果との一致解析結果。
Figure2B:一致したデータセットで、WESベースのSMNコーラーから取得したSMN1コピー数コールとWGSベースのSMNコーラーから取得したSMN1コピー数コールの一致解析結果。
Figure2C:一致したデータセットで、WESベースのSMNコーラーから取得したSMN2コピー数コールとWGSベースのSMNコーラーから取得したSMN2コピー数コールの一致解析結果
外部臨床コホートの結果
スペインのJuno Geneticsは、独立した部分的盲検化(SMNでは665/1117、HBAでは464/1099の盲検化)とIllumina DNA Prep with Exome 2.5 EnrichmentとIllumina CS/PGx Custom Enrichment Research Panelスパイクインの追加(1009のユニークサンプルから1140データセット)でプロファイリングされた臨床サンプルの部分的連続コホートを生成し、IlluminaはDRAGEN v4.4でコホートを解析しました。Junoは、自動化システムを用いてエクソームライブラリーを調製し、NovaSeq 6000でシーケンスし、各S4フローセルに192サンプルまたはS2フローセルに96サンプルのいずれかをロードしました。このコホートのほとんどのサンプルは、HBA遺伝子座のMLPAプロファイリングと、エクソームベースのコールの精度を評価するために使用されたSMN1コピー数のqPCRまたはMLPAプロファイリングの結果と一致していました。QCに合格したサンプルにおける、一致するMLPAまたはqPCR検査によるエクソーム結果の一致率は、HBAでは100%(1099/1099)、SMN1コピー数およびSMAステータス(1113/1115)では99.8%でした。このコホートのバッチ内の他のサンプルとの深度プロファイルの不一致によりQCに失敗したサンプルはありません。この臨床サンプルコホート(100%)と、開発に使用された細胞株コホートイルミナ(94.1%)の間でQCに合格したサンプルの割合が有意に高いことから、自動化システムで調製された臨床サンプルでは、異なるオペレーターによって手動で調製された細胞株よりも、より堅牢で一貫した結果が得られることが示されています。
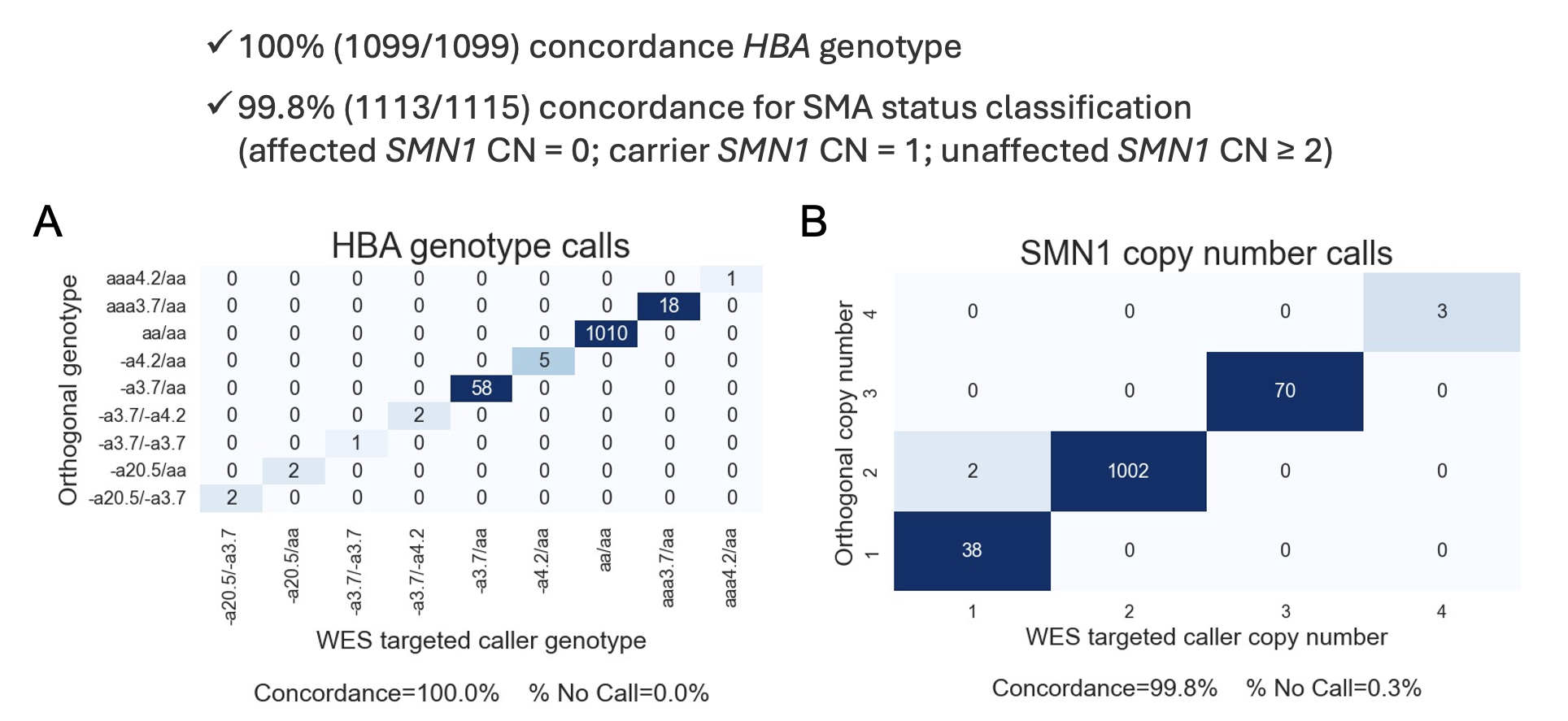
図1A:臨床サンプルコホートにおけるWESベースのHBAコーラーと直交アッセイ(MLPA)結果の一致解析結果。
Figure3B:WESベースのSMNコーラーから得られたSMN1コピー数コールと臨床サンプルコホートの直交アッセイ(qPCR)結果との一致解析結果。
結論
イルミナDNA Prep with Exome 2.5 Enrichmentに追加された、DRAGENおよびイルミナCS/PGx Custom Enrichment Research Panelで実装された新しいエクソームベースのHBAおよびSMNターゲットコーラーは、統合されたNGSワークフロー内でHBAおよびSMN遺伝子をプロファイリングするための信頼できるアプローチを示しています。 Illumina DNA Prep この方法は、キャリアスクリーニング研究における直交アッセイへの依存を減らし、費用対効果を高め、そのようなデータセットのアクセシビリティを向上させる可能性があります。