ペアエンドシーケンスとシングルリードシーケンスの比較
ペアエンドシーケンスとシングルリードシーケンスの違いは何ですか?
シングルリードシーケンスでは、DNAを一端のみからシーケンスします。これは、イルミナのシーケンスを利用する最もシンプルな方法です。シングルリードシーケンスとは異なり、ペアエンドシーケンスでは、断片の両端をシーケンスするため、アライメントしやすい高品質なシーケンスデータを生成できます。このため、ゲノム再編成や反復配列要素に加え、遺伝子融合や新規転写産物の検出が容易になります。
また、ライブラリー調製にかかる時間と労力は同じであっても、取得できるリード数は2倍となります。さらに、シーケンスがリードペアとしてアライメントされることで、リードアライメントの精度が向上し、シングルリードデータでは検出が難しい挿入欠失(Indel)バリアントの検出も可能です1。ペアエンドシーケンスは、イルミナのすべての次世代シーケンサー(NGS)システムで実施できます。
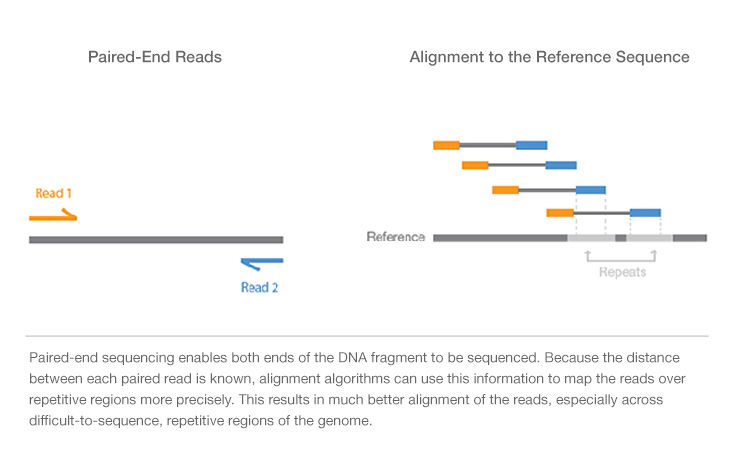
ペアエンドシーケンスとシングルエンドシーケンスの利点の比較
ペアエンドシーケンス
- シンプルなペアエンドライブラリー:シンプルなワークフローにより、多様な範囲のインサートサイズを生成できます。
- 効率的なサンプル使用:シングルリードゲノムDNAまたはcDNAシーケンスと同量のDNAが必要です。
- 幅広いアプリケーション:DNAのメチル化や制限消化が不要で、バイサルファイトシーケンシングに使用できます。
- シンプルなデータ解析: ショートインサートライブラリーを用いた高品質のシーケンスアセンブリを実現します。標準的なシングルリードライブラリー調製プロセスに簡単な変更を加えることで、各クラスターの順鎖テンプレートと逆鎖テンプレートの両方を、1回のペアエンドリードシーケンスで読み取ることができます。いずれのリードにもロングレンジの位置情報が含まれるため、リードの高精度なアライメントが可能です。
シングルリードシーケンス
- コスト効率の高い用途:本ソリューションは、高品質データを迅速かつ経済的に大量に提供します。
- 特定のアプリケーション:シングルリードシーケンスは、Small RNA-Seqやクロマチン免疫沈降シーケンス(ChIP-Seq)などの特定の手法に適しています。
ゲノムとトランスクリプトミクスのペアエンドシーケンス
ペアエンドDNAシーケンス
ペアエンドDNAシーケンスリードは、反復シーケンスを含むDNA領域全体で高品質のアライメントを提供し、コンセンサス配列のギャップを埋めることで、de novoシーケンス用の長いコンティグを生成します。ペアエンドDNAシーケンスは、挿入、欠失、反転などの一般的なDNA再構成も検出します。
ペアエンドRNAシーケンス
ペアエンドRNAシーケンス(RNA-Seq)は、がんにおける遺伝子融合の検出や、新規スプライスアイソフォームの特性解析などの発見アプリケーションを可能にします。2
イルミナでは、ペアエンドRNA-Seq向けに、代替の断片化プロトコールを用いたキットを提供しています。これに続き、標準的なイルミナのペアエンドクラスター形成とシーケンスが実施されます。

遺伝子発現と制御研究による影響力の高い発見
RNAシーケンス技術に加え、シングルセルRNA-Seqや空間RNA-Seq、タンパク質、クロマチン、メチル化解析などの補完的技術が、生物学および疾患の理解にどのような影響をもたらしているかをご覧ください。
関連コンテンツ
次世代シーケンサーテクノロジー
NGSで実行できる幅広い実験について理解を深めるとともに、主要なシーケンシングテクノロジーの進歩をご覧ください。
NGSシーケンスによるChIPアッセイ
ChIP-Seqを使用して、偏りのないゲノムワイドな遺伝子制御の洞察を得る方法をご確認ください。
全エクソームシーケンス
ゲノムのタンパク質コード領域に焦点を当て、エクソンバリアントを検出することで、疾患や公衆衛生に対する遺伝的影響の解明に貢献します。
教育ウェビナー
全ゲノムシーケンスにおけるアプリケーションと進歩
全ゲノムシーケンスにおける最新の進歩と、新規のゲノミクステクノロジーの大きな可能性についてご覧ください。
がん研究におけるNGSの再定義
がん研究者が、どのようにNGSを使用してプロテオーム、エピゲノム、ノンコーディングRNA、さらには低分子RNAの特性評価を行っているかをご覧ください。
サンプルからゲノムの洞察まで
ゲノムの重要な洞察を得るためのイルミナのソリューションについて、Mile Lelivelt(VP of Product Management for Software and Informatics)の見解をご紹介します。
注目の製品
Illumina Stranded mRNA Prep
コーディングトランスクリプトーム解析に適した、シンプルかつスケーラブルで費用対効果が高く、1日で完了できる迅速なソリューションです。
Illumina Stranded Total RNA Prep with Ribo-Zero Plus
コーディングおよびノンコーディングトランスクリプトーム研究に対応する多様なサンプルタイプから迅速にライブラリーを調製できるため、高い柔軟性を持って研究を進めることが可能です。
シーケンシングの実施方法についてさらに知るには:
補足資料
Illumina Sequencing by Synthesisのメカニズム
Illumina Sequencing by Synthesis(SBS)ケミストリーの仕組みをご覧ください。

イルミナシーケンスプラットフォーム
ベンチトップおよび生産規模のシーケンサーを確認し、適切なプラットフォームの選択に役立つリソースをご利用ください。

発現解析における短いペアエンドリードと長いシングルエンドリードの比較
本論文では、RNA-Seqにおいて、長いシングルエンドリードと比較した短いペアエンドリードの利点を説明しています。

ライブラリー調製
革新的かつ包括的なライブラリー調製ソリューションは、イルミナのシーケンスワークフローを支える重要な要素です。
参考文献
- Nakazato T, Ohta T, Bono H. Experimental design-based functional mining and characterization of high-throughput sequencing data in the sequence read archive. PLoS One. 2013;8(10):e77910.
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63.