遺伝子解析
遺伝子解析とは?
遺伝子解析は、DNAサンプルの研究を記述して、個人の疾患リスクを増加させたり、薬物応答に影響を与えたりする可能性のある違いやバリアントを調べるために使用される用語です。
ハンチントン病のような一部の遺伝性疾患は単一の遺伝子変化が原因ですが、ほとんどの遺伝性疾患は環境要因によって悪化する可能性のある多くの遺伝子変化から生じ、研究者が関連するすべての要因を追跡することは困難です。
遺伝子レベルでは99.9%が同一ですが、0.1%の違いは、ある人が別の人よりも特定の疾患にかかりやすい理由を説明するのに役立ちます。遺伝的変異のパターンを研究することは、どの違いが疾患に関連するかを決定するのに役立ちます。
遺伝的変異と疾患生物学を理解する
ヒトゲノムやさまざまなモデル生物のシーケンスに、幅広いリソースが投資されています。現在、遺伝的変異と機能に対する理解を深め、これらの知見を利用して将来の新しい診断法や治療法を開発するために、さらなるリソースが提供されています。
主要な疾患の基盤を理解するために、膨大な数の遺伝子マーカーと患者サンプルを解析する必要性が引き続きあります。イルミナのテクノロジーは、これらの研究での使用に特有な立場にあり、DNA、RNA、タンパク質レベルでの遺伝的変異の理解を深めるのに役立ちます。
DNA、RNA、タンパク質の基本
ヒトの体は数十億の細胞から成り立っており、それぞれが細胞機能の基本的指示をコードするデオキシリボ核酸(DNA)を含んでいます。生物のDNAの完全なセットをゲノムと呼びます。
ヒトゲノムは23対の染色体に編成され、さらに遺伝子と呼ばれる30,000を超える小さな領域に分割されます。各遺伝子は、A、C、G、Tとラベルされた一連のヌクレオチド塩基で構成されています。ヒトDNAには約30億のヌクレオチド塩基があり、その正確な順序はDNAシーケンスとして知られています。
遺伝子が"発現すると、"DNAシーケンスの部分コピーであるメッセンジャーRNA(リボ核酸)またはmRNAが、タンパク質の合成を導くテンプレートとして使用されます。
タンパク質は次に、すべての細胞機能を誘導します。
SNPとは?
一塩基多型(SNP)は、DNAにおける一塩基(A、G、C、T)の変化です。SNPは、遺伝的変異の最も多い原因です。ヒトゲノムには1,000万を超えるSNPが含まれていると考えられており、300塩基に約1塩基含まれています。
SNPを調べるための多くの遺伝子解析ツールが利用できます。研究者は、ゲノム全体にわたる数十万のSNPを解析し、健康な患者サンプルと疾患のある患者のサンプル間のパターンを比較することができます。フォローアップのファインマッピング研究では、特定の疾患の原因SNPを見つけるために、ゲノムの定義された領域内の少数のSNPの解析を行う場合があります。
遺伝子発現とは?
遺伝子発現解析は、特定の細胞または細胞群においてどの遺伝子が活性であるかを決定するプロセスです。一般的に、遺伝子とタンパク質の中間であるメッセンジャーリボ核酸(mRNA)を測定することで研究されます。
正常組織と疾患組織など、異なる環境の細胞間で遺伝子発現パターンを比較することで、研究者はさまざまな疾患状態においてどの遺伝子が活性または不活性であるかを判断できます。
DNAメチル化とは?
DNAメチル化は、ゲノムのCpG部位のシトシンヌクレオチドへのメチル基の結合です。DNAメチル化は遺伝子転写制御に影響を与えると考えられており、遺伝性の可能性があります。
DNAメチル化の失敗は、がん、糖尿病、および特定の神経疾患を含むさまざまなヒト疾患と関連しているため、メチル化パターンは疾患バイオマーカーとして使用される可能性があります。
Copy Number Variation(CNV)とは?
CNVとは、リファレンスゲノムと比較した場合の領域のDNAコピー数レベルの変化を指します。最近の研究では、ヒトゲノムのCNVレベルは、これまで考えられていたよりもはるかに高いことがわかっています。
他の種類のコピー数の変化は、染色体内または染色体全体の領域が不規則に増幅されたり、完全に削除されたりした場合に起こります。コピー数異常はがん細胞に典型的なものです。一部の腫瘍では、高レベルの増幅が臨床転帰と相関します。
ヘテロ接合性喪失(LOH)とは?
LOHは、親が細胞のゲノムの一部に関与していないことを意味します。LOHはがんにおいて非常に一般的であり、多くの場合、がん領域に腫瘍抑制遺伝子が存在することを示しています。通常、腫瘍抑制遺伝子の残りのコピーは点変異によって不活化されます。
LOHは、明らかなコピー数の変化(遺伝子変換など)なしに発生することがあり、最近の論文では腫瘍サンプルにおけるコピー中立LOHの重要な役割が文書化されています。このようなイベントは、従来の2色マイクロアレイ比較ゲノムハイブリダイゼーション(アレイCGH)では検出できません。
イルミナのDNA解析ソリューションにより、研究者は、LOHのゲノムだけでなく、がんやその他の疾患で日常的に見られるその他の染色体異常を、より高い分解能でプロファイリングすることができます。
ゲノミクスニュース

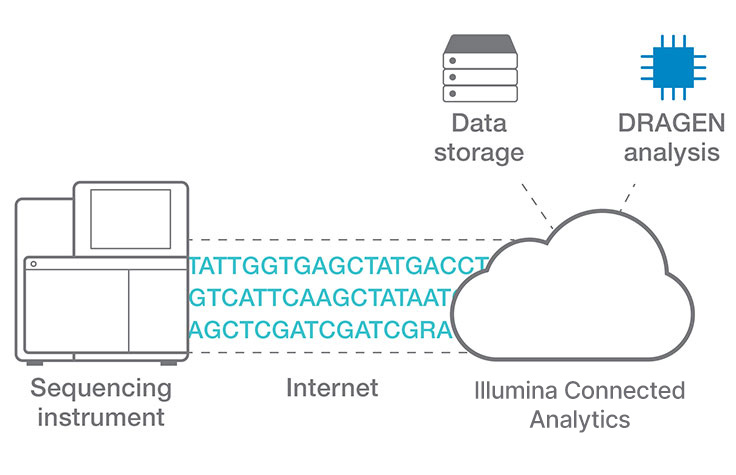
COPDマルチオミクスプロジェクトのための効率的なクラウドデータ解析
日本の岡山大学の研究者らによる、Illumina Connected AnalyticsとDRAGENパイプラインを使用した、全ゲノム、エクソーム、トランスクリプトーム、メタゲノムデータ解析
インタビューを読む
Can Applying NGS Lead to Better Beer?
An international group of scientists, yeast producers, and brewers sequenced 96 yeast strains to learn how genetics can influence the flavor of beer. Their findings, published in Cell, could help brewers make better beers.
Read Article