NGS用語ガイド
シーケンスプロジェクトを計画する際に、重要な用語の定義を確認し重要な概念を明確にするのに役立つ次世代シーケンサー用語集です。
サンガーシーケンスとNGSの重要な違いはシーケンス量です。サンガー法では一度に1つのDNA断片のみをシーケンスしますが、NGSは極めて並行です。つまり、ランごとに数百万の断片を同時にシーケンスします。このプロセスにより、一度に数百から数千の遺伝子をシーケンスします。また、NGSには、ディープシーケンスで新規または希少なバリアントを検出するより高い検出力もあります。
一般的なNGS用語の用語集
NGSライブラリー調製の一環として、シーケンスライブラリー内の各DNA断片の5’末端と3’末端にライゲーションされた短い配列特異的オリゴ。アダプターは、イルミナフローセルの表面に存在する短いシーケンスを補完するものです。
シーケンスリードをリファレンスゲノムに一致させるプロセス。
イルミナフローセルの表面で発生する増幅反応。フローセルの製造中、表面は「P5」および「P7」と呼ばれる2つの異なるオリゴヌクレオチドの芝生でコーティングされます。ブリッジ増幅の最初のステップでは、一本鎖シーケンスライブラリー(相補的なアダプターエンド付き)がフローセルにロードされます。ライブラリー内の個々の分子は、オリゴの芝生を「流れ」ながら相補的なオリゴに結合します。プライミングは、ライゲーションされた断片の反対側の端が曲がり、表面上の別の相補的なオリゴに「架橋」した時に起こります。(PCRに似た)変性と伸長サイクルを繰り返すと、単一分子がフローセル全体で数百万のユニークでクローン性のクラスターに局所的に増幅されます。この「クラスター」とも呼ばれるプロセスは、NGS装置内のオンボードクラスターモジュールで発生します。
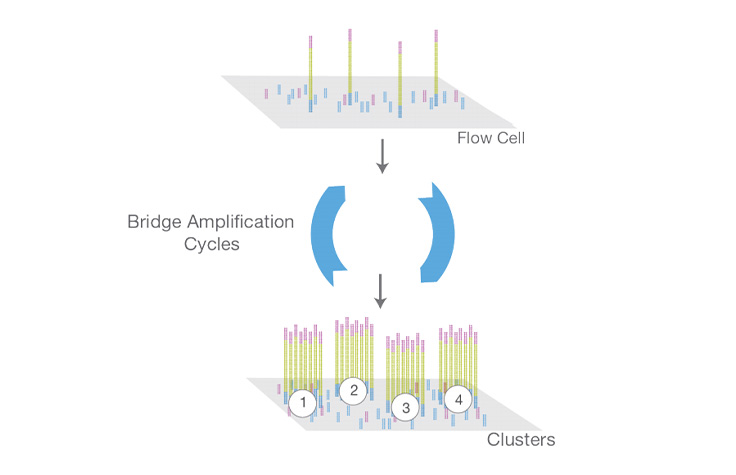
クラスター増幅
ライブラリーはフローセルにロードされ、断片はフローセル表面にハイブリダイズされます。各結合断片は、ブリッジ増幅を通じてクローンクラスターに増幅されます。
フローセルの表面に結合したテンプレートDNAのクローングループ。各クラスターは単一のテンプレートDNA鎖で播種され、ブリッジ増幅を通じてクラスターが約1000コピーになるまでクローン的に増幅されます。フローセル上の各クラスターは、単一のシーケンスリードを生成します。例えば、フローセル上の10,000クラスターでは、10,000シングルリードと20,000ペアエンドリードが生成されます。
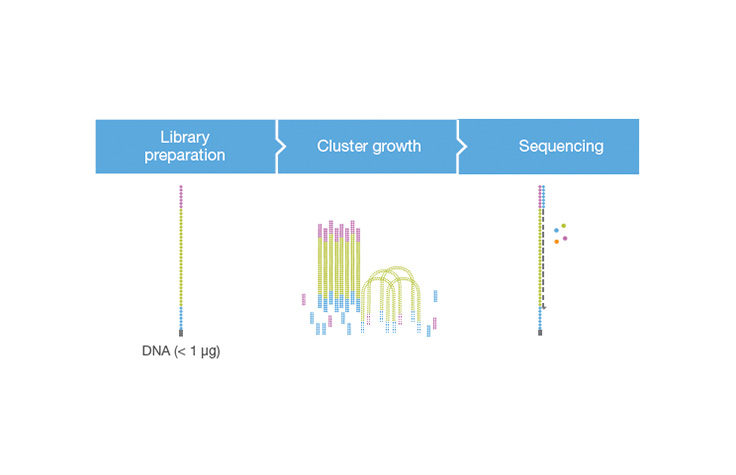
クラスター
各i5インデックスがマトリックス内の各i7インデックスとペアになり、固有のインデックスペアを作成し、固有の片面インデックスは作成しないインデックスペアです。
シーケンスリードのオーバーラップをアラインすることで生成される連続シーケンスのin silico。
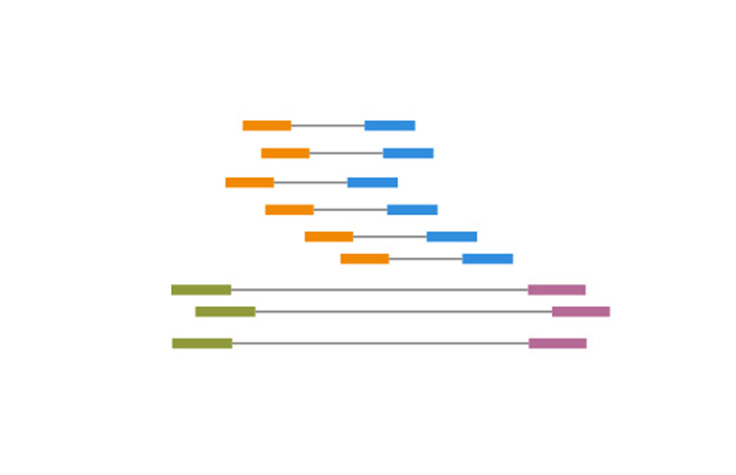
コンティグ
既知のリファレンス塩基にアラインする、または「カバー」するシーケンスされた塩基の平均数。例えば、30×カバレッジでシーケンスされた全ゲノムは、ゲノムの各塩基が平均30回シーケンスされたことを意味します。より高いカバレッジレベルでは、ベースコールはより高い信頼度で行うことができます。
詳細はこちら所定の深度でゲノムまたはターゲット領域を横切ってシーケンスされた塩基の割合を示すメトリクス(例:95%の塩基が10倍以上のカバレッジでカバーされている)。平均シーケンス深度(例:30×平均カバレッジ)だけでは、許容限界値未満でシーケンスされた塩基の割合や、全くシーケンスされなかった塩基の割合は考慮されません。例えば、データセットの"カバレッジ分布が95%で、カバレッジが10倍以上の場合、"これは塩基の5%が10×閾値未満でカバーされたか、またはまったくカバーされなかったことも示しています。このため、シーケンス結果を説明するために、一般的にカバレッジ分布が平均カバレッジとともに使用されます。
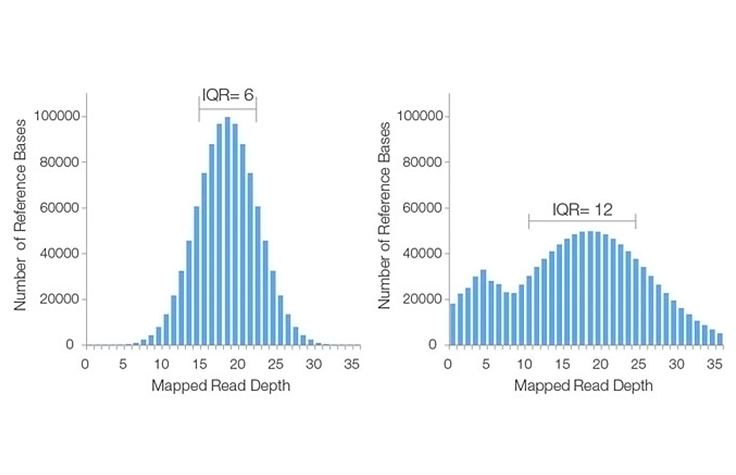
カバレッジの分布
NGS装置の消耗品として使用される、1、2、または8レーンが物理的に分離されたガラススライドまたはその他の固体表面。シーケンステンプレートはフローセル表面に固定化されており、表面結合テンプレートの高い安定性と蛍光標識ヌクレオチドの低い非特異的結合を保証しながら、酵素へのアクセスを助ける形でDNAを提示するように設計されています。固相増幅(クラスター形成)は、近接する各単一テンプレート分子の同一コピーを最大1,000個作成します。1平方センチメートルあたり約1,000万クラスターの密度を達成します。

ライブラリー調製中に各DNA断片に添加されるユニークな短いDNAシーケンス。ユニークなシーケンスにより、多くのライブラリーを一緒にプールし、同時にシーケンスすることができます。プールされたライブラリーからのシーケンスリードは、最終的なデータ解析の前に、そのバーコードに基づいて同定され、計算的にソートされます。ライブラリーマルチプレックスは、小さなゲノムや関心のあるゲノム領域をターゲットにする場合に有用な手法です。バーコードでマルチプレックスを行うことにより、コストやランタイムの大幅な増加なしに、1回のランで分析できるサンプル数を飛躍的に増やせます。
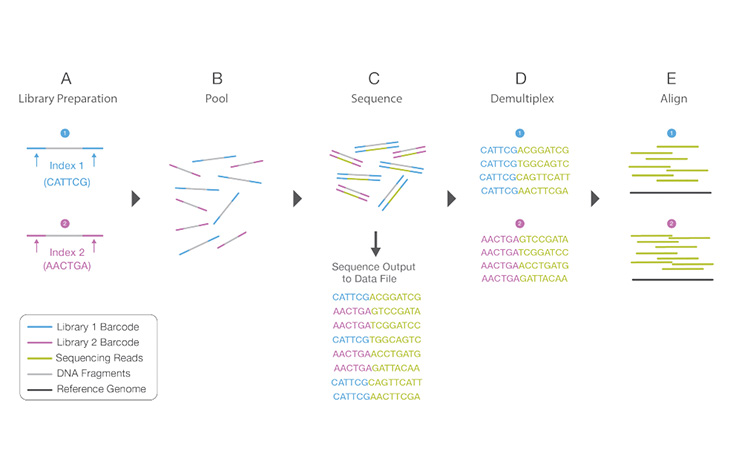
ライブラリーマルチプレックスの概要
(A)ユニークなインデックスシーケンスが、ライブラリー調製中に2つの異なるライブラリーに添加されます。(B)ライブラリーが緒にプールされ、同じフローセルレーンにロードされます。(C)ライブラリーが、1回の装置ラン中に一緒にシーケンスされます。すべてのシーケンスが単一の出力ファイルにエクスポートされます。(D) デマルチプレックス法のアルゴリズムが、インデックスに従ってリードを異なるファイルにソートします。(E)リードの各セットが適切なリファレンスシーケンスにアライメントされます。
i5およびi7シーケンスは、イルミナシーケンスランのインデックスリードで、関心のあるシーケンスを含むR1およびR2リードから上流(i5)および下流(i7)にあります。短いアダプターをヌクレオチド断片に添加することで、イルミナのシーケンサーとの互換性を確保します。これらのシーケンスは、シーケンサーのフローセル上の相補的シーケンスに結合します。
ライブラリー調製中、サンプルDNAは断片化され、特定のサイズの断片(通常は200~500 bpですが、より大きい場合もあります)が2つのオリゴアダプターの間にライゲーションまたは「挿入」されます。元のサンプルDNA断片は「インサート」とも呼ばれます。
ゲノムDNAサンプル(またはcDNAサンプル)をシーケンスライブラリーに変換する分子生物学プロトコールで、NGS装置でシーケンスすることができます。ライブラリー調製の最初のステップは、DNAサンプルのランダムな断片化です。その後、各DNA断片に5’アダプターと3’アダプターをライゲーションします。あるいは、「タグメンテーション」により断片化反応とライゲーション反応を1つのステップにまとめ、ライブラリー調製プロセスの効率を大幅に向上させます。
詳細はこちら次世代シーケンサーの別名。
ライブラリー調製中に、固有の短いDNAシーケンス、すなわち「インデックス」を各DNA断片に添加するプロセス。ユニークなシーケンスにより、多くのライブラリーを一緒にプールし、同時にシーケンスすることができます。プールされたライブラリーからのシーケンスリードは、最終データ解析の前に同定され、計算的にソートされます。ライブラリーマルチプレックスは、小さなゲノムや関心のあるゲノム領域をターゲットにする場合に有用な手法です。マルチプレックスにより、コストやランタイムの大幅な増加なしに、1回のランで分析できるサンプル数を飛躍的に増やせます。
詳細はこちら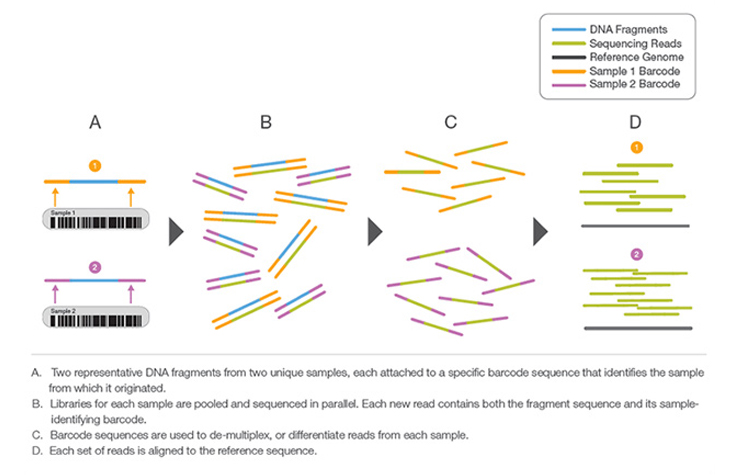
マルチプレックス
シーケンスライブラリー内のすべてのDNA断片にわたる各塩基位置に、A、C、G、Tヌクレオチドの等比率を有すること。イルミナのシーケンスシステムで効果的なイメージ解析を行うには、カラーバランスが必要です。したがって、ほとんどのイルミナライブラリー調製ワークフローにはランダムな断片化ステップが含まれており、ライブラリーの各塩基位置で必要なシーケンスの多様性を生成します。
イルミナの知識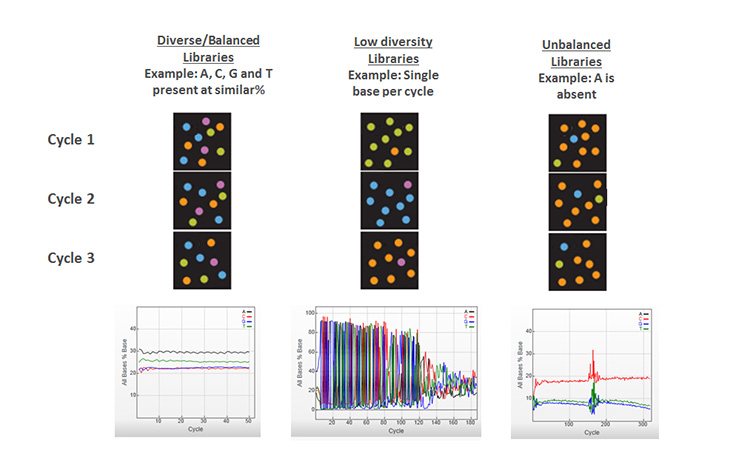
ヌクレオチドの多様性
短いDNAまたはRNAシーケンス。
同じランでDNA断片の両端からシーケンスするプロセス。
詳細はこちら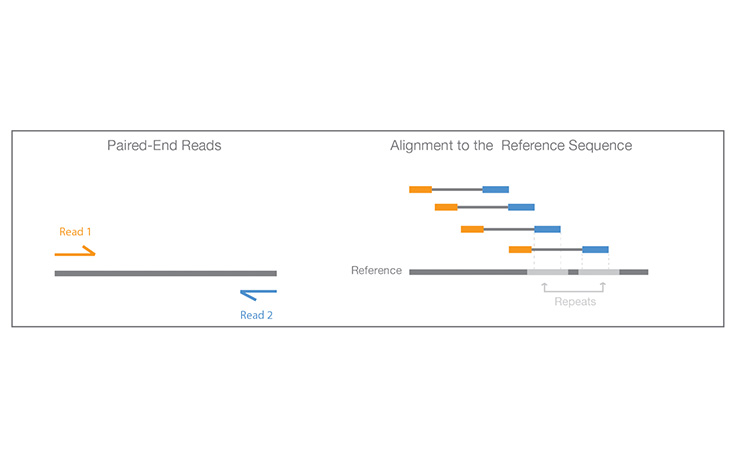
ペアエンドシーケンスとアライメント
ペアエンドシーケンスでは、DNA断片の両端をシーケンスします。各ペアリード間の距離は既知であるため、この情報をアライメントアルゴリズムに反映させることで、反復領域のリードをより正確にマッピングできます。その結果、リードのアライメント効率が改善されます。これは、シーケンスが困難なゲノムの反復領域間で特に有効です。
NGSのメトリクスで、ベースコーリングにおけるエラーの確度を予測または推定します。クオリティスコア(Qスコア)は、非常に小さなエラー確率を伝達するコンパクトな方法です。高いQスコアは、ベースコールの信頼性が高く、不正確である可能性が低いことを意味します。
詳細はこちら1回の"リード"は、イルミナフローセルのブリッジ増幅プロセスで得られたヌクレオチド断片の順鎖および逆鎖のシーケンスを指します。
リファレンスゲノムは、新しいシーケンスリードがアライメントされ比較されるスキャフォールドとして機能する、完全にシーケンスされ組み立てられたゲノムです。通常、シーケンスランから生成されたリードは、データ解析の最初のステップとしてリファレンスゲノムにアライメントされます。リファレンスゲノムの例には、hg19およびhg38が含まれます。
SBSテクノロジーは、蛍光標識された4つのヌクレオチドを使用して、フローセル表面上の数千万のクラスターを並行してシーケンスします。各シーケンスサイクル中に、単一標識デオキシヌクレオチド三リン酸(dNTP)が核酸鎖に添加されます。ヌクレオチドの標識は重合化の「可逆ターミネーター」として機能します。dNTPの取り込み後、蛍光色素はレーザーの励起とイメージングによって識別され、酵素的に切断されて次回の取り込みを可能にします。ベースコールは、各サイクル中のシグナル強度測定から直接行われます。4つの可逆性ターミネーター結合dNTP(A、C、T、G)はすべて、単一かつ個別の分子として存在するため、自然競合は、組み込みバイアスを最小限に抑えます。

Sequence by Synthesis(SBS)テクノロジー
特定のサンプル内の特定のバリアントを検出する能力を指します。アリル頻度が低いほど、検出に必要な感度も高くなります。次世代シーケンサーは、毛細血管電気泳動よりも高い感度を提供し、希少な変異を検出する能力を提供します。
シーケンスリードの開始点を示すアダプターシーケンスに隣接するPCRプライマー。シーケンスプロセス中、プライマーはテンプレートストランド上のシーケンスアダプターの一部にアニールします。DNAポリメラーゼ酵素がこの部位に結合し、相補的ヌクレオチド塩基を成長する反対鎖に、塩基ごとに組み込みます。
シーケンスライブラリーの各DNA断片に対応するA、T、C、G塩基のデータ文字列。イルミナのテクノロジーでは、ライブラリーがシーケンスされると、各DNA断片がフローセルの表面上にクラスターを生成し、各クラスターが単一のシーケンスリードを生成します。(例えば、フローセル上の100万クラスターでは、100万のシングルリードと200万のペアエンドリードが生成されます。) リード長は、アプリケーションのニーズに応じて25 bp~300 bp以上の範囲になります。
基質に付着する蛍光色素の明るさ。Sequence by Synthesisでは、ベースコールは各サイクル中のシグナル強度測定から直接行われます。
イルミナはシングルインデックスとデュアルインデックスを含むいくつかのインデックス手法をサポートしています。シングルインデックスでは、最大48の固有の6塩基インデックスを使用して、最大48の固有にタグ付けされたライブラリーを生成できます。デュアルインデックスでは、最大24の固有の8塩基インデックス1シーケンスと最大16のユニーク8塩基インデックス2シーケンスを組み合わせて使用し、最大384の固有にタグ付けされたライブラリーを生成できます。
二本鎖DNAが同時に断片化され、イルミナのアダプター配列とPCRプライマー結合部位でタグ付けされる迅速な酵素反応。混合反応により、ライブラリー調製中の個別の機械的断片化ステップが不要になります。
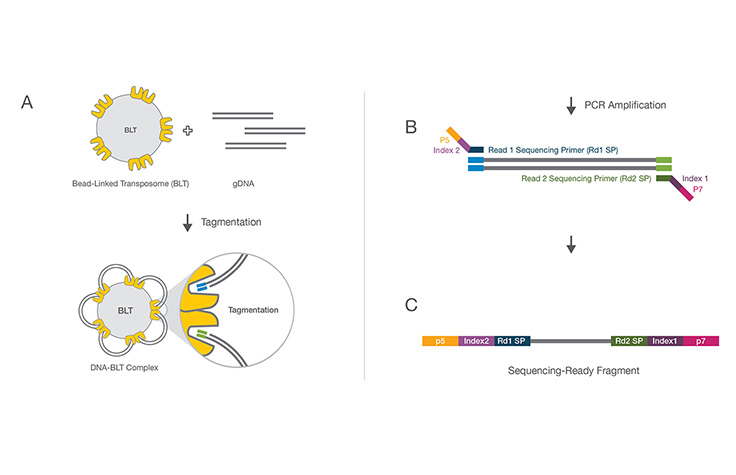
タグメンテーション
ターゲットリシーケンス実験におけるすべてのターゲット領域の合計サイズ。ターゲットサイズは、事前に設計されたパネルやカスタムパネルによって異なります。(例えば、22のターゲット領域のパネルでは、各ターゲット領域が100 kbであり、合計ターゲットサイズは2200 kbになります。)
次世代シーケンサー装置によって生成されるデータ量。通常、メガベース(Mb)またはギガベース(Gb)で定義されます。
1メガ塩基=1,000,000塩基。
1ギガ塩基=1,000,000,000塩基。
1メガ塩基=1,000,000塩基。
1ギガ塩基=1,000,000,000塩基。
すべてのi5インデックスとすべてのi7インデックスが1回のみ使用されるようなインデックスのペア。ユニークデュアルインデックスにより、インデックスホッピングのリードを同定してフィルタリングできるため、マルチプレックスサンプルの信頼性が向上します。
記事を読むゲノムのタンパク質コード領域(エクソーム)のみをターゲットとする広く使用されているシーケンス法。
詳細はこちら
NGSの導入eBook
ご研究へのNGSの導入をお考えですか? 新しいテクノロジーの採用には不安がつきまとうものです。NGSをラボに導入するための包括的でわかりやすいガイドを作成しました。NGS手法、ワークフロー、データ解析ソリューション、追加の用語集用語について学び、NGSを始めるためのステップバイステップガイドを見つけることができます。
eBookをダウンロードする新着NGS eBookをダウンロードする
補足資料
初心者向けNGS
NGSについての詳細にご関心をお持ちですか? このページでは、次世代シーケンサーとその従来の手法に勝る多くの利点について、簡潔で明確にご説明します。
NGSチュートリアル
NGSの主な概念を理解するのに役立つチュートリアルを多数ご用意しています。ライブラリー調製、シーケンス、およびデータ解析のベストプラクティスについて、順を追って説明します。
オンライントレーニング
装置、ライブラリー調製キット、ソフトウェアなどのオンデマンドトレーニングにアクセスできます。